HI-VQE Chemistry — функція Qiskit від Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
See the API reference
Функції Qiskit є експериментальною можливістю, доступною лише користувачам IBM Quantum® Premium Plan, Flex Plan та On-Prem (через IBM Quantum Platform API) Plan. Вони перебувають у статусі попереднього випуску і можуть змінюватися.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Огляд
У квантовій хімії задача електронної структури полягає у знаходженні розв'язків електронного рівняння Шрьодінґера — квантових хвильових функцій, що описують поведінку електронів системи. Ці хвильові функції є векторами комплексних амплітуд, де кожна амплітуда відповідає внеску можливої конфігурації електронів.
Основний стан — це хвильова функція системи з найнижчою енергією, яка має особливе значення при вивченні молекулярних систем. Найточніший підхід до обчислення основного стану враховує всі можливі конфігурації електронів, але він стає неможливим для більших систем, оскільки кількість конфігурацій зростає експоненційно зі збільшенням розміру системи.
Ітераційний варіаційний квантовий власний солвер з передачею (HI-VQE) — це інноваційний гібридний квантово-класичний метод для точного оцінювання основного стану молекулярних систем. Він інтегрує квантове апаратне забезпечення з класичними обчисленнями, використовуючи квантові процесори для ефективного дослідження кандидатних конфігурацій електронів та обчислення відповідної хвильової функції на класичних комп'ютерах. Генеруючи компактні, але хімічно точні хвильові функції, HI-VQE сприяє дослідженням і відкриттям у квантовій хімії та матеріалознавстві.

HI-VQE зменшує обчислювальну складність задачі електронної структури, ефективно оцінюючи основний стан з високою точністю. Він зосереджується на ретельно відібраній підмножині найбільш релевантних конфігурацій електронів, оптимізуючи як точність, так і ефективність.
Поєднуючи переваги класичних і квантових комп'ютерів, HI-VQE ітераційно вдосконалює й покращує поточну оцінку хвильової функції. Його унікальні техніки побудови підпростору допомагають зробити вибір конфігурацій ефективнішим, надаючи користувачам більший обчислювальний контроль та покращену точність у квантово-хімічних симуляціях.
Якщо хочеш детальніше дізнатися про алгоритм, можеш прочитати відповідну дослідницьку статтю.
Опис
Кількість конфігурацій електронів для молекулярної системи зростає експоненційно зі збільшенням розміру системи. Однак для певних електронних станів, таких як основний стан, зазвичай лише невелика частка конфігурацій суттєво впливає на енергію стану. Методи вибраної конфігураційної взаємодії (SCI) використовують цю розрідженість для зменшення обчислювальних витрат, ідентифікуючи та зосереджуючись на найбільш релевантних конфігураціях. Ця підмножина конфігурацій називається підпростором.
HI-VQE використовує притаманну квантовим комп'ютерам ефективність при поданні молекулярних систем для допомоги в пошуку підпростору. Він інтегрує класичні та квантові підпрограми для розв'язання задачі електронної структури з високою точністю. На відміну від існуючих квантових SCI-методів, HI-VQE поєднує варіаційне навчання, ітераційну побудову підпростору та попереднє діагоналізаційне відсіювання конфігурацій для підвищення ефективності шляхом зменшення кількості квантових вимірювань, ітерацій і витрат на класичну діагоналізацію. Тому HI-VQE можна застосовувати до більших молекулярних систем, які вимагають більше кубітів, і зменшує вартість розв'язання задачі заданого розміру до однакового ступеня точності.
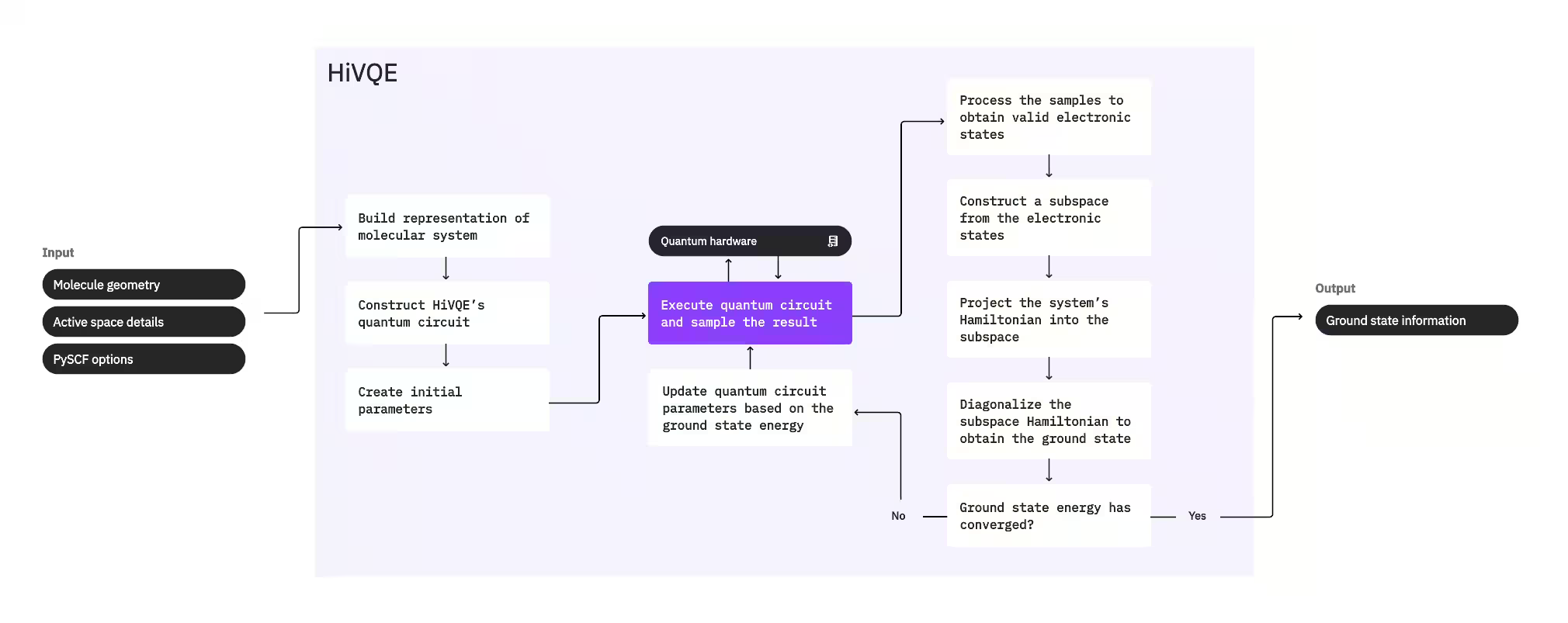
Для обчислення основного стану системи HI-VQE спочатку використовує класичний хімічний пакет PySCF для генерації молекулярного представлення з наданих користувачем вхідних даних, таких як молекулярна геометрія та інша молекулярна інформація. Потім він входить у гібридний квантово-класичний цикл оптимізації, ітераційно вдосконалюючи підпростір для оптимального представлення основного стану, мінімізуючи кількість включених конфігурацій. Цикл продовжується до виконання критеріїв збіжності, таких як розмір підпростору або стабільність енергії, після чого виводяться обчислена хвильова функція основного стану та енергія. Ці результати можна використовувати для побудови точних поверхонь потенційної енергії та проведення подальшого аналізу системи.
Цикл оптимізації зосереджується на налаштуванні параметрів квантової схеми для генерації якісного підпростору. HI-VQE пропонує три варіанти квантових схем: excitation_preserving, efficient_su2 та LUCJ. Оптимізація ініціалізується поблизу референсного стану Гартрі–Фока завдяки його загальній придатності. Потім схема виконується на квантовому пристрої, і конфігурації вибираються з отриманого квантового стану перед поверненням у вигляді бінарних рядків. Через шуми квантового пристрою деякі відібрані конфігурації можуть бути фізично недійсними, не зберігаючи кількість електронів або спін. HI-VQE вирішує це за допомогою процесу відновлення конфігурацій з пакету qiskit-addon-sqd, щоб користувачі могли або виправити недійсні конфігурації, або відкинути їх.
Дійсні конфігурації потім проходять необов'язковий крок відсіювання для видалення тих, що, за прогнозами, дають мінімальний внесок. Це зменшує розмірність підпростору, тим самим знижуючи вартість кроку діагоналізації. Якщо відсіювання увімкнено, то будується попередній підпросторовий гамільтоніан із дійсних конфігурацій і виконується діагоналізація з дуже м'якими критеріями завершення. Хоча точність отриманих амплітуд для кожної конфігурації є низькою, це ефективно для прогнозування, які конфігурації слід виключити з підпростору на даній ітерації, і обчислюється швидко.
Вибрані конфігурації додаються до підпростору, і гамільтоніан системи проєктується у цей підпростір. Підпростір оновлюється ітераційно, зберігаючи найбільш релевантні конфігурації між ітераціями. Цей підхід відрізняється від альтернативних методів тим, що квантова схема не потребує апроксимації повного основного стану на кожному кроці.
Далі підпросторовий гамільтоніан класично діагоналізується для отримання найменшого власного значення та відповідного власного вектора, що представляють апроксимацію основного стану та його енергії. З покращенням якості підпростору через ітерації обчислений основний стан краще апроксимує істинний основний стан. На цьому етапі може виконуватися додатковий крок відсіювання для видалення з підпростору конфігурацій, що не мають суттєвого внеску в обчислений основний стан. Цей крок забезпечує максимальну компактність підпростору, що переноситься до наступної ітерації. Це оцінюється на основі амплітуд, що повертаються діагоналізацією, оскільки вони представляють важливість внеску кожної конфігурації в обчислений основний стан.
Перевірка збіжності потім визначає, чи подальше навчання покращить результати. Якщо так, виконується необов'язковий крок класичного розширення, параметри квантової схеми оновлюються для подальшої мінімізації обчисленої енергії, і процес повторюється. Крок класичного розширення генерує додаткові конфігурації для підпростору, доповнюючи конфігурації, відібрані з квантового пристрою. Спочатку він визначає конфігурацію з найбільшою амплітудою в результатах діагоналізації, а потім генерує нові конфігурації з одиничними та подвійними збудженнями від визначеної конфігурації. Потрібна кількість цих конфігурацій потім додається до підпростору.
Як тільки визначається, що ітерації збіглись, HI-VQE повертає обчислений основний стан (у формі станів у підпросторі та їхніх амплітуд у хвильовій функції основного стану), його енергію та міру дисперсії енергії, що дає уявлення про те, чи є обчислений стан власним станом гамільтоніана системи.
Користувачі можуть вибирати квантову схему та кількість знімків для кожної квантової схеми, а також контролювати розмір підпростору або вмикати класичну генерацію додаткових конфігурацій на допомогу конфігураціям, згенерованим квантовим способом. Таким чином користувачі можуть налаштовувати поведінку HI-VQE відповідно до своїх цільових застосувань.
Ліцензування
Зверни увагу, що використання цієї функції Qiskit обмежено задачами, що вимагають не більше 20 кубітів, якщо не отримано ліцензію, яка надає вищий ліміт.
Будь ласка, надішли електронного листа на адресу qiskit.support@qunovacomputing.com, якщо хочеш дізнатися про отримання ліцензії.
Початок роботи
Спочатку запроси доступ до функції. Потім автентифікуйся за допомогою свого API-ключа IBM Quantum® і, якщо ти вже зберіг свій обліковий запис у локальному середовищі, вибери функцію Qiskit таким чином:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Приклад
Перший приклад показує, як обчислити енергію основного стану молекули NH3 за допомогою алгоритму HI-VQE.
Визначення молекулярної геометрії та параметрів
Молекулярна геометрія NH3 задається декартовими координатами, розділеними символом ";" для кожного атома.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Додаткові параметри можна визначити та надати для молекулярної системи у наступному форматі словника.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Виконай функцію з вхідними даними геометрії та параметрів.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Варто роздрукувати ідентифікатор завдання функції, щоб його можна було надати у запитах на підтримку у разі виникнення проблем.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Цей приклад використовує 16 кубітів з 8 орбіталями базису sto3g для молекули NH3. Перевіряй статус свого навантаження функції Qiskit або отримуй результати таким чином:
print(job.status())
QUEUED
Після завершення завдання результати можна отримати за допомогою екземпляра result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Для доступу до енергії основного стану використовуй ключ "energy". Ключ "eigenvector" надає CI-коефіцієнти з відповідними позначеннями конфігурацій електронів у вигляді бітових рядків, збережених у "states" результатів.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Продуктивність
У цьому розділі наведено продемонстровані еталонні обчислення HI-VQE для випадку з 24 кубітами для Li2S, випадку з 40 кубітами для молекули N2 та випадку з 44 кубітами для системи FeP-NO.
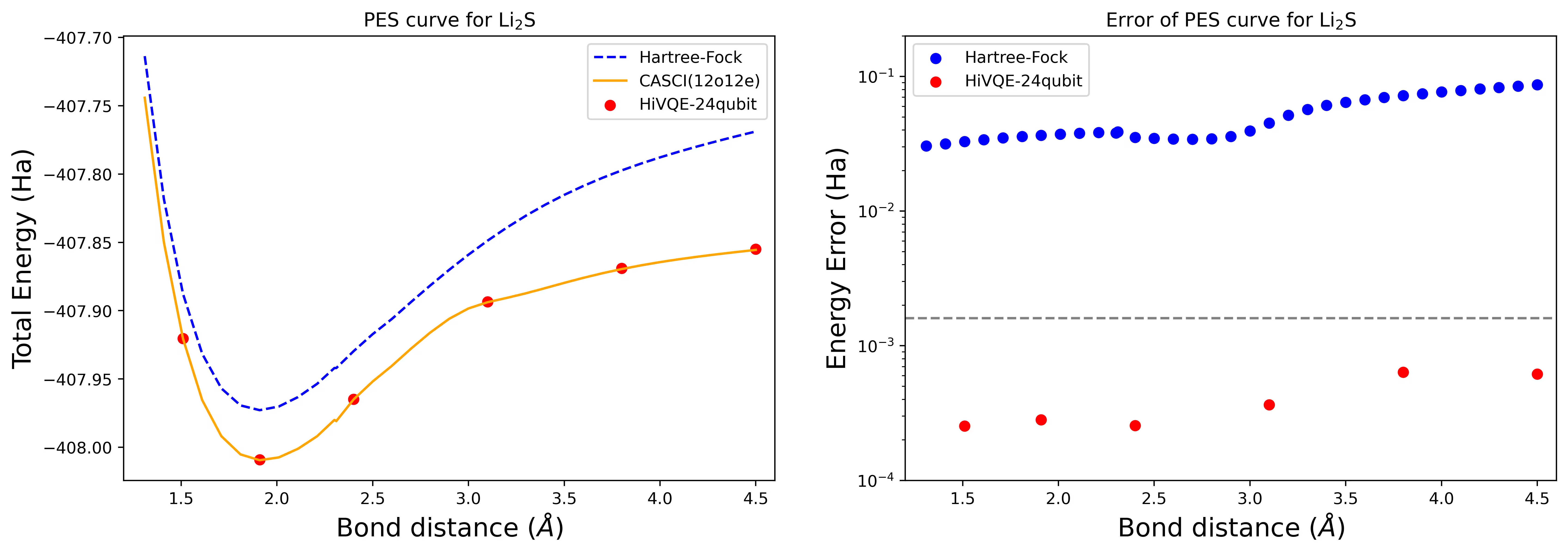
Крива поверхні дисоціативної потенційної енергії для молекули Li2S з 24 кубітами
Крива PES показана з референсом FCI та початковим наближенням RHF, а також похибкою енергії від референсу FCI.
 .
.
Обчислення проводилися з такими геометріями та параметрами.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Червоні точки представляють результати розрахунків HI-VQE для шести різних геометрій, а три геометрії, що відповідають 1.51, 2.40 та 3.80 ангстремам, надані як вхідні дані у наведеній вище комірці.
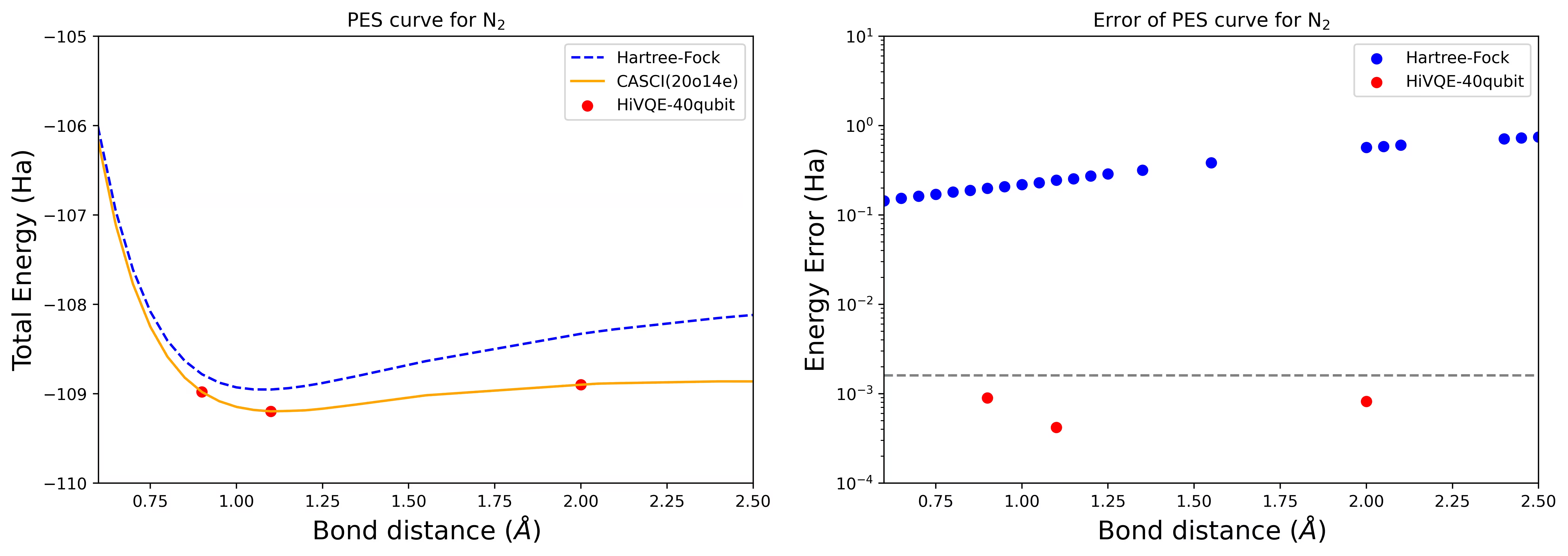
Крива дисоціативної PES для молекули N2 з 40 кубітами
Молекула азоту ідентифікована як мультиреференсна система з великими внесками кореляційної енергії, що виходять за межі стану Гартрі–Фока. Ми провели еталонні обчислення для молекули N2 з базисом cc-pvdz, (20o,14e) з використанням вибору активних орбіталей homo-lumo. Число повного активного простору (CAS) для представлення цієї задачі становить 6 009 350 400. Неможливо отримати розв'язок задачі на власні значення (для енергії та електронної структури) з такою кількістю станів на потужному настільному комп'ютері (16 ЦП/64 ГБ). За допомогою HI-VQE користувачі можуть ефективно шукати в підпросторі станів CAS для отримання хімічно точних результатів при значній економії обчислювальних ресурсів. Наступні графіки показують криву PES 40-кубітних обчислень HI-VQE для дисоціації молекули N2.
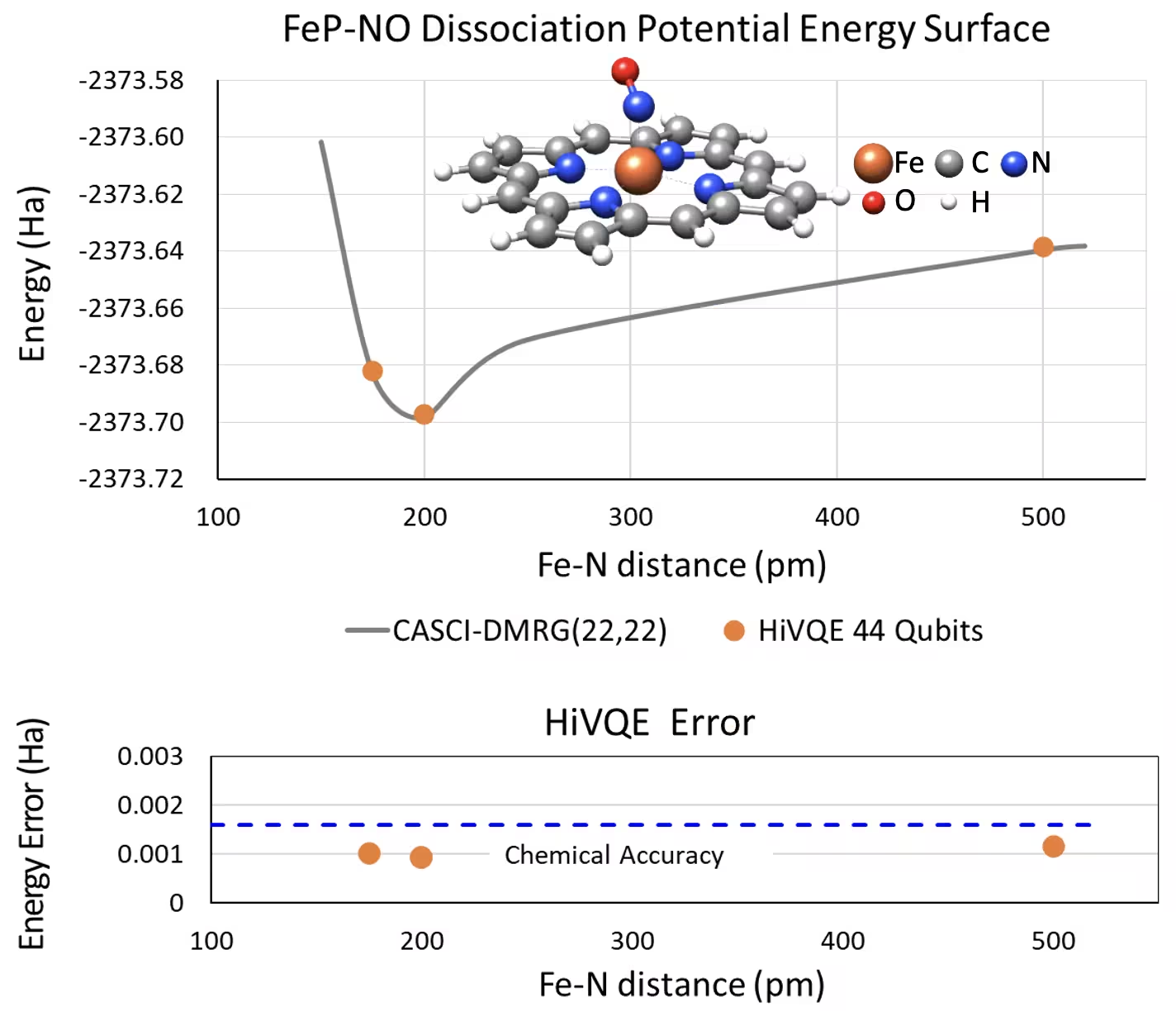
Крива дисоціативної PES для п'ятикоординованого заліза(II)-порфірину з системою NO з 44 кубітами
Ще одна цікава хімічна система — це комплекс заліза(II)-порфірину (FeP) з координованим лігандом монооксиду азоту (NO), який являє собою біологічно значиму металопорфіринову систему, що відіграє ключову роль у різних фізіологічних процесах. У цьому прикладі HI-VQE використовувався для оцінки точної кривої поверхні потенційної енергії міжмолекулярної взаємодії між FeP та NO (енергія основного стану для геометрій з різними відстанями). Комбінована система має 450 орбіталей і 202 електрони (450o,202e) з базисом 6-31g(d) в цілому. Вибір активних орбіталей homo-lumo використовувався для обчислення меншого випадку від реального з (22o,22e). З наступних еталонних результатів нам вдалося досягти хімічної точності (> 1.6 мГа) порівняно з найсучаснішим класичним хімічним обчисленням CASCI(DMRG) (22o,22e) як референсом.
Еталонні показники
- Точний розмір матриці — це кількість детермінантів для точного розв'язку, наприклад FCI та CASCI.
- Обчислення HI-VQE вибирає та обчислює підпростір (тобто розмір матриці HI-VQE).
- Загальний час включає час виконання на QPU та запуски функції Qiskit з CPU.
- Точність оцінюється за різницею енергії від точного розв'язку.
| Хімічна система | Кількість кубітів | Точний розмір матриці | Розмір матриці HI-VQE | E(diff) від точного (мГа) | Кількість ітерацій | Загальний час | Використання QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0.08 | 6 | 37 с | 34 с |
| (10o,10e) | 20 | 63504 | 3969 | 0.60 | 5 | 250 с | 50 с |
| (15o,10e) | 30 | 9018009 | 49729 | 0.90 | 5 | 354 с | 54 с |
| (16o,14e) | 32 | 130873600 | 1798281 | 1.10 | 9 | 6531 с | 121 с |
| (18o,24e) | 36 | 344622096 | 399424 | 0.90 | 24 | 5174 с | 130 с |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1.20 | 21 | 46547 с | 258 с |
Отримання повідомлень про помилки
Якщо твоє навантаження завершиться з помилкою, статус буде ERROR, а виклик job.result() викличе виключення:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Отримати підтримку
Ти можеш надіслати електронного листа на адресу qiskit.support@qunovacomputing.com для отримання допомоги з цією функцією.
Якщо тобі потрібна допомога з усуненням конкретної помилки, будь ласка, надай ідентифікатор завдання функції для завдання, що зіткнулось з помилкою.
Наступні кроки
- Запроси доступ до функції, заповнивши цю форму.
- Відвідай довідник API для цієї функції Qiskit.
- Спробуй посібник Обчислення кривої дисоціативної PES для FeP-NO за допомогою HI-VQE.
- Ознайомся з Pellow-Jarman, A., та ін. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Спробуй посібник Криві дисоціативної PES з Qunova HiVQE.