Варіаційний квантовий розв'язувач власних значень (VQE)
Для цього модуля студенти повинні мати налаштоване середовище Python та встановлені останні версії таких пакетів:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Щоб налаштувати та встановити ці пакети, звернись до посібника Встановлення Qiskit. Щоб запускати завдання на реальних квантових комп'ютерах, студенти повинні створити обліковий запис IBM Cloud, виконавши кроки з посібника Налаштування облікового запису IBM Cloud.
Цей модуль було протестовано і використало приблизно 8 хвилин часу QPU. Це приблизна оцінка, і твоє фактичне використання може відрізнятися.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Вступ
З часів розвитку квантово-механічної моделі на початку XX століття вчені розуміють, що електрони не рухаються по фіксованих траєкторіях навколо ядра атома, а існують у просторових областях імовірності, які називаються орбіталями. Ці орбіталі відповідають конкретним дискретним рівням енергії, які можуть займати електрони. Електрони природно перебувають на найнижчих доступних рівнях енергії — так званому основному стані. Однак якщо електрон поглинає достатньо енергії, він може перейти на вищий рівень, потрапивши у збуджений стан. Цей збуджений стан є тимчасовим, і врешті-решт електрон повертається на нижчий рівень енергії, вивільняючи поглинуту енергію, часто у вигляді світла. Цей фундаментальний процес поглинання та випромінювання енергії є важливим для розуміння того, як атоми взаємодіють і утворюють хімічні зв'язки.
Коли атоми з'єднуються, утворюючи молекули, їхні атомні орбіталі комбінуються, формуючи молекулярні орбіталі. Розташування та рівні енергії електронів у цих молекулярних орбіталях визначають властивості молекули, що утворюється, та міцність хімічних зв'язків. Наприклад, при утворенні молекули водню () з двох окремих атомів водню електрон кожного атома займає атомні орбіталі. По мірі того як атоми зближуються, їхні атомні орбіталі перекриваються і комбінуються, утворюючи нові молекулярні орбіталі — одну з нижчою енергією (зв'язуюча орбіталь) і одну з вищою енергією (розпушуюча орбіталь). Два електрони, по одному від кожного атома водню, переважно займають зв'язуючу орбіталь з нижчою енергією, що призводить до утворення стабільного ковалентного зв'язку, який утримує молекулу . Різниця в енергії між розділеними атомами і молекулою, що утворилась, зокрема енергія електронів у молекулярних орбіталях, визначає стабільність і властивості зв'язку.
У наступних розділах ми розглянемо цей процес утворення молекул, зосередившись на молекулі . Ми використаємо реальний квантовий комп'ютер разом із класичними методами оптимізації, щоб знайти енергію цього простого, але фундаментального процесу. Цей експеримент стане практичною демонстрацією того, як квантові обчислення можна застосувати для вирішення задач обчислювальної хімії, розкриваючи роль електронної енергії.
VQE — варіаційний квантовий алгоритм для задач на власні значення
Методи наближення в хімії — варіаційний принцип і базисний набір
Внесок Ервіна Шредінгера в квантову механіку не обмежується введенням нової електронної моделі; по суті, він заклав основи хвильової механіки, розробивши знамените залежне від часу рівняння Шредінгера:
Тут — оператор Гамільтоніана, який представляє повну енергію системи, а — хвильова функція, що містить усю інформацію про квантовий стан системи. (Примітка: — повна похідна за часом, і ми явно не включаємо сюди власне значення енергії .)
Однак у багатьох практичних застосуваннях — наприклад, при визначенні дозволених рівнів енергії атомів і молекул — ми натомість використовуємо незалежне від часу рівняння Шредінгера (рівняння власних значень енергії), яке виводиться з залежного від часу рівняння за припущення стаціонарного стану. Стаціонарний стан — це квантовий стан, у якому щільність імовірності знаходження частинки в заданій точці простору не змінюється з часом.
У цьому вигляді представляє власне значення енергії, що відповідає квантовому стану . Гамільтоніан включає різні внески до енергії: кінетичну енергію електронів і ядер, сили притягання між електронами і ядрами, а також сили відштовхування між електронами.
Розв'язання рівняння власних значень енергії дає нам змогу обчислити квантовані рівні енергії атомних і молекулярних систем. Однак для молекул точне розв'язання є складним, оскільки хвильова функція , що описує просторовий розподіл електронів, має складний і багатовимірний характер.
Тому вчені використовують методи наближення для отримання практичних і точних розв'язків. У цій роботі ми зосередимося на двох ключових підходах:
-
Варіаційний принцип
Цей метод наближає хвильову функцію і коригує її, щоб максимально наблизитися до цільової енергії — зазвичай до енергії основного стану системи. Ключова ідея варіаційного принципу проста:
- Якщо ми підбираємо хвильову функцію ("пробну функцію"), обчислена з неї енергія завжди буде рівна або більша за енергію основного стану () системи.
- Підбираючи параметри пробної функції, , можна дедалі точніше наближати енергію основного стану.
- Точність цього методу сильно залежить від вибору пробної хвильової функції . Погано підібрана пробна функція може призвести до оцінки енергії, далекої від точної.
-
Наближення базисного набору
Другий метод наближення застосовується на етапі побудови хвильової функції — підхід базисного набору. У квантовій хімії точне розв'язання рівняння Шредінгера для молекул є практично неможливим. Замість цього ми апроксимуємо складну багатоелектронну хвильову функцію, будуючи її з простіших, заздалегідь визначених математичних функцій. Базисний набір — це, по суті, колекція таких відомих математичних функцій, зазвичай зосереджених навколо атомів у молекулі, які слугують будівельними блоками для представлення форми та поведінки електронів у системі. Уяви, що намагаєшся відтворити деталізовану скульптуру, використовуючи лише набір стандартних деталей LEGO — чим більше типів і розмірів деталей у тебе є (чим більший базисний набір), тим точніше можна наблизити оригінальну форму.
Ці базисні функції часто натхненні аналітичними розв'язками для простих систем, як-от атом водню, і набувають вигляду функцій Гаусса або типу Слейтера, хоча й залишаються наближеннями. Замість роботи з теоретично "точними", але практично нездійсненними повними молекулярними орбіталями, ми виражаємо їх як лінійну комбінацію (суму з коефіцієнтами) цих базисних функцій. Цей підхід відомий як метод лінійної комбінації атомних орбіталей (ЛКАО), коли базисні функції нагадують атомні орбіталі. Оптимізуючи коефіцієнти цієї лінійної комбінації, можна знайти найкращу можливу наближену хвильову функцію і енергію в межах обраного базисного набору.
- Що більше функцій включено до базисного набору, то краще наближення, але це потребує більших обчислювальних ресурсів.
- Малий базисний набір дає грубу оцінку, тоді як великий базисний набір забезпечує точніші результати ціною більших обчислювальних витрат.
Підсумовуючи: щоб зробити обчислення здійсненними і знизити їх вартість, ми застосовуємо варіаційний принцип, наближаючи хвильову функцію — це зменшує обчислювальну складність і дозволяє ітеративно оптимізувати для мінімізації енергії. Водночас підхід базисного набору спрощує обчислення, представляючи атомні орбіталі як комбінацію заздалегідь визначених функцій, замість того щоб розв'язувати задачу для неперервної хвильової функції безпосередньо.
Перевір своє розуміння
Розглянь пробну хвильову функцію , де — константа нормування, а — регульований параметр.
(a) Нормуй пробну хвильову функцію, визначивши так, щоб .
Answer
Для нормування заданої пробної хвильової функції:
Використовуємо інтеграл Гаусса:
підставляємо і отримуємо:
(b) Обчисли очікуване значення Гамільтоніана , заданого виразом де , що відповідає потенціалу простого гармонічного осцилятора.
Answer
Гамільтоніан гармонічного осцилятора:
Очікуване значення кінетичної енергії
Беремо другу похідну:
Отже:
Використовуючи стандартні результати для інтегралів Гаусса:
Очікуване значення потенційної енергії
Використовуючи:
отримуємо:
Повне очікуване значення енергії
(c) Використай варіаційний принцип, щоб знайти оптимальне , мінімізуючи .
Answer
Оптимізуємо для мінімальної енергії
Диференціюємо
Розв'язуємо:
Підставляємо у :
що збігається з точною енергією основного стану квантового гармонічного осцилятора.
VQE (Варіаційний квантовий розв'язувач власних значень)
Варіаційний квантовий розв'язувач власних значень (VQE) — це основний метод, який ми використаємо для дослідження процесу . Давай спочатку розберемося, що таке VQE і як він працює. Але спершу зупинимося на одній дуже важливій речі через контрольне запитання.
Перевір своє розуміння
Якщо у нас вже є так багато стратегій для хімічних задач, то навіщо нам потрібен квантовий комп'ютер? І яка мета спільного використання квантових і класичних комп'ютерів?
Answer
Квантові обчислення мають шанс революціонізувати хімію, вирішуючи проблеми, які класичним комп'ютерам важко подолати через експоненційне масштабування квантових станів. Річард Фейнман свого часу зауважив, що для моделювання природи обчислення теж повинні бути квантовими [ref 1].
Наприклад, моделювання кофеїну з найпростішим базисним набором (STO-3G) потребувало б бітів — це значно більше, ніж загальна кількість зірок у видимому Всесвіті () [ref 2]. Квантовий комп'ютер здатний описати електронні орбіталі кофеїну за допомогою 160 кубітів.
Квантові комп'ютери природним чином обробляють квантові взаємодії, використовуючи суперпозицію та заплутаність, що відкриває перспективний шлях до точного молекулярного моделювання. До того ж ми можемо поєднати переваги квантових комп'ютерів (моделювання електронів) і класичних (попередня/наступна обробка даних, управління алгоритмічними процесами, оптимізація тощо). Очікується, що це прискорить відкриття нових матеріалів, розробку ліків і передбачення реакцій, зменшуючи кількість дорогих експериментів методом спроб і помилок. [ref 3][ref 4]
Якщо ти хочеш дізнатися більше про те, навіщо квантові комп'ютери потрібні для хімічних задач і чому варто використовувати як квантові, так і класичні обчислювальні ресурси, ознайомся з такими статтями:
Тепер повернемося до VQE.
VQE поєднує потужність квантових і класичних комп'ютерів, принципово спираючись на варіаційні принципи для визначення енергії основного стану системи. Щоб зрозуміти VQE, розбий його на три частини:
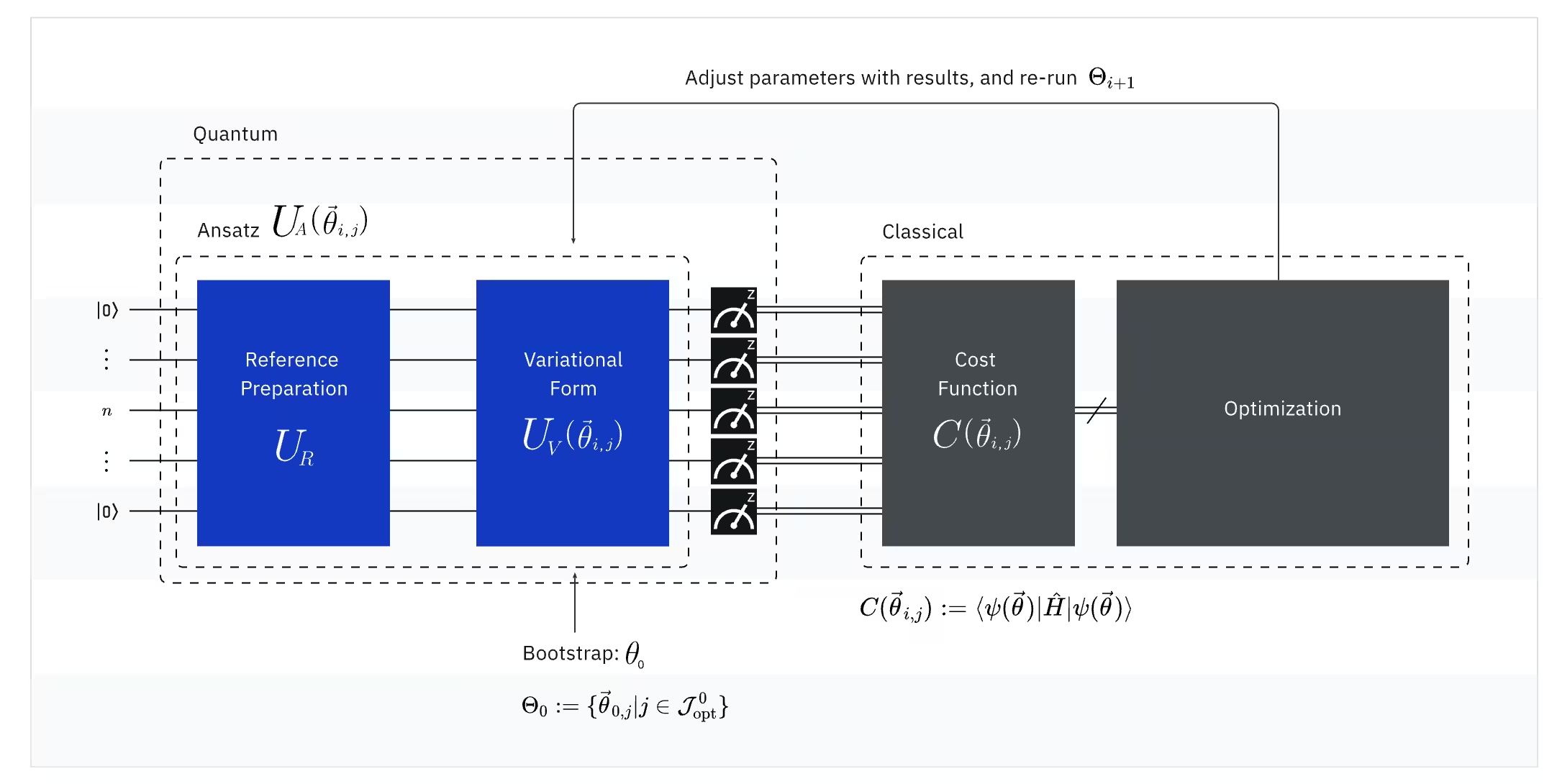
(Квантовий) Спостережуваний: молекулярний Гамільтоніан (енергія молекули)
У VQE молекулярний/атомний Гамільтоніан є спостережуваним, тобто ми можемо виміряти його значення в ході експерименту. Наша мета — знайти найнижчу можливу енергію (енергію основного стану) молекули. Для цього ми використовуємо пробний квантовий стан, що генерується параметризованою квантовою схемою (анзац). Ми вимірюємо спостережуваний і оптимізуємо квантовий стан, доки не досягнемо найнижчої можливої енергії.
Базисний набір, що використовується для молекулярного Гамільтоніана, визначає необхідну кількість кубітів і безпосередньо впливає на точність VQE. Правильний вибір базисного набору є критично важливим для балансу між ефективністю та точністю. Щоб спростити обчислення без зміни базисного набору, можна використовувати такі стратегії, як врахування симетрії та редукція активного простору. Багато молекул мають симетричну форму (наприклад, метелик або сніжинка), що означає: деякі їхні частини поводяться однаково. Замість того щоб обчислювати все окремо, можна зосередитися лише на унікальних частинах, заощаджуючи квантові ресурси завдяки симетрії. При редукції активного простору враховуються лише важливі орбіталі, оскільки не всі електрони суттєво впливають на молекулярну енергію. Електрони поблизу ядра здебільшого залишаються незмінними, тоді як інші відіграють роль у формуванні зв'язків. Застосовуючи ці методи, можна зробити VQE ефективнішим, не жертвуючи точністю.
Отримавши молекулярний Гамільтоніан із відповідним базисним набором і використовуючи описані вище стратегії, нам потрібно перетворити цей Гамільтоніан у форму, придатну для квантових комп'ютерів. Відображення задач на оператори Паулі може бути досить складним — особливо в квантовій хімії, де доводиться мати справу з нерозрізнюваними частинками (електронами), тоді як кубіти є розрізнюваними. Ми не будемо тут заглиблюватися в деталі відображень, але звернемо тебе до таких ресурсів. Загальний опис відображення задачі на квантові оператори можна знайти в курсі Quantum computing in practice. Детальніший розгляд відображення хімічних задач на квантові оператори — у курсі Quantum chemistry with VQE.
Для цього модуля ми надамо тобі відповідні (однокубітні) Гамільтоніани для і , щоб ти міг зосередитися на роботі з квантовим комп'ютером. Ці однокубітні Гамільтоніани підготовлені з використанням базисного набору STO-6G та відображення Джордана-Вігнера — найбільш прямолінійного відображення з найпростішою фізичною інтерпретацією, оскільки воно відображає заповнення однієї спін-орбіталі на заповнення одного кубіту. Також було застосовано техніку редукції кубітів із використанням симетрії Гамільтоніана, яка використовує закономірності в поведінці спінових заповнень для зменшення кількості кубітів. Для молекули ми припускаємо, що відстань між двома атомами водню дорівнює 0.735 .
(Квантовий) Анзац: пробна хвильова функція (як побудувати пробний квантовий стан за допомогою квантової схеми)
Для VQE анзац (множина: ansätze) складається з двох ключових компонентів. Перший — це підготовка початкового стану, яка встановлює стан кубіту шляхом застосування квантових гейтів без варіаційних параметрів. Другий компонент — параметризована квантова схема: спеціальна квантова схема з регульованими параметрами, подібними до ручок налаштування на радіоприймачі. Ці параметри використовуватимуться в останній частині — класичному оптимізаторі — щоб досягти найкращого можливого основного стану.
У розділі про варіаційний принцип ми дізналися, що якість пробного стану впливає на якість результатів варіаційного алгоритму. Це означає, що вибір хорошого анзацу є важливим у VQE. Ця тема є глибокою і складною. Ми не будемо тут розглядати різні типи анзаців та їхнє походження. Якщо тебе цікавить більше інформації про параметризовані квантові схеми та анзац, ти можеш ознайомитися з уроком Ansatz and variational form з курсу Variational algorithm design course, який містить детальні пояснення та приклади ansätze.
Оскільки в цьому модулі ми будемо використовувати однокубітний Гамільтоніан, нам потрібна однокубітна параметризована квантова схема у якості анзацу. Далі ми розглянемо три типи однокубітних ansätze, порівняємо їх і обговоримо ключові аспекти вибору анзацу.
(Класичний) Оптимізатор: тонке налаштування квантової схеми
Після того як квантовий комп'ютер вимірює енергію спостережуваного з анзацу, параметри анзацу та значення енергії надсилаються до класичного оптимізатора для налаштування. Цей процес оптимізації виконується на класичному комп'ютері, зазвичай із використанням загальнонаукових пакетів, як-от SciPy.
Класичний оптимізатор розглядає виміряну енергію як цільову функцію. В задачах оптимізації цільова функція (яку також іноді називають функцією вартості) — це математична функція, що вимірює "якість" конкретного розв'язку. Мета оптимізатора — знайти набір параметрів, який мінімізує цю цільову функцію. У контексті пошуку енергії основного стану молекули сама по собі енергія слугує цільовою функцією — ми хочемо знайти параметри для нашої квантової схеми (наш "розв'язок"), які дають найнижчу можливу енергію. Класичний оптимізатор використовує це виміряне значення енергії (вартість) і визначає наступний набір оптимізованих параметрів для квантового анзацу. Ці оновлені параметри потім повертаються назад до квантової схеми, і процес повторюється. З кожною ітерацією класичний оптимізатор регулює параметри, намагаючись знизити енергію (мінімізувати цільову функцію), доки не буде виконано заздалегідь визначений критерій збіжності — в ідеалі це гарантує знаходження найнижчої можливої енергії (що відповідає основному стану молекули для даної відстані між атомами і базисного набору).
Наукові пакети, як-от SciPy, пропонують багато стратегій оптимізації. Більше про це можна дізнатися в уроці Optimization loops курсу Variational algorithm design. Тут ми використаємо COBYLA (Constrained Optimization BY Linear Approximations) — алгоритм оптимізації, придатний для складних енергетичних ландшафтів. Зокрема, COBYLA не намагається обчислити градієнт досліджуваної функції — такий підхід називається оптимізацією без обчислення градієнта. Уяви, що намагаєшся знайти найвищу вершину в гірському хребті із заплющеними очима. Оскільки ти не бачиш усього ландшафту, ти робиш маленькі кроки в різних напрямках, перевіряючи, чи йдеш вгору чи вниз. COBYLA працює аналогічно — він рухається по просторі параметрів, тестуючи різні значення, поступово покращуючи результат, поки не знайде найкращий.
Тепер ти готовий виконати обчислення VQE. Для цього спробуй контрольне запитання нижче, яке підсумовує весь процес.
Перевір своє розуміння
Заповни пропуски правильними термінами, щоб доповнити зведення процесу VQE, а потім натисни, щоб перевірити відповіді.
VQE — це варіаційний квантовий алгоритм, який поєднує потужність (1) ________ і класичних обчислень, призначений для знаходження (2) __________ молекули. Процес починається з визначення (3) __________, що представляє повну енергію системи і виступає спостережуваним у квантових вимірюваннях. Далі ми підготовляємо (4) __________ — квантову схему з регульованими параметрами, яка представляє пробну хвильову функцію молекули. Ці параметри оптимізуються за допомогою (5) __________ — класичного алгоритму, що ітеративно коригує параметри для мінімізації виміряної енергії. В описаному вище ми використали оптимізатор (6) __________, який уточнює параметри анзацу без потреби в обчисленні похідних. Процес продовжується, доки не буде досягнуто (7) __________, тобто поки ми не знайдемо найнижчу можливу енергію молекули.
Банк слів:
- класичний оптимізатор
- енергія основного стану
- hardware-efficient
- анзац
- молекулярний Гамільтоніан
- COBYLA
- квантові обчислення
- збіжність
Answer
1 → квантові обчислення
2 → енергія основного стану
3 → молекулярний Гамільтоніан
4 → анзац
5 → класичний оптимізатор
6 → COBYLA
7 → збіжність
Обчислення енергії основного стану атома водню за допомогою VQE
Тепер застосуємо вивчене для обчислення енергії основного стану атома водню. Протягом усього модуля ми будемо використовувати фреймворк для квантових обчислень під назвою "Qiskit patterns", який розбиває робочі процеси на такі кроки:
- Крок 1: Відображення класичних вхідних даних на квантову задачу
- Крок 2: Оптимізація задачі для квантового виконання
- Крок 3: Виконання з використанням примітивів Qiskit Runtime
- Крок 4: Постобробка та класичний аналіз

Ми загалом будемо дотримуватися цих кроків.
Почнемо із завантаження необхідних пакетів, включаючи примітиви Qiskit Runtime. Ми також виберемо найменш завантажений доступний квантовий комп'ютер.
Нижче є код для збереження облікових даних під час першого використання. Після збереження облікових даних у своєму середовищі обов'язково видали цю інформацію з ноутбука, щоб вони випадково не були розкриті під час його поширення. Дивись посібники Set up your IBM Cloud account та Initialize the service in an untrusted environment для отримання докладнішої інформації.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Клітинка нижче дозволить тобі переключатися між використанням симулятора та реального апаратного забезпечення протягом усього ноутбука. Рекомендуємо запустити її зараз:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Крок 1: Відображення задачі на квантові схеми та оператори
Ми починаємо обчислення VQE з визначення Гамільтоніана для молекули водню () при конкретній відстані між атомами. Цей Гамільтоніан представляє повну енергію системи у вигляді кубітних операторів і був отриманий та відображений із молекулярної системи за допомогою стандартної процедури: 1) застосування базисного набору STO-6G (конкретна колекція математичних функцій для наближення електронних орбіталей), 2) застосування відображення Джордана-Вігнера (техніка для перетворення ферміонних операторів, що описують електрони, на кубітні оператори) та 3) виконання редукції кубітів із використанням симетрій Гамільтоніана для спрощення задачі.
Як ми вже пояснювали, обчислені енергії основного стану суттєво залежать від вибору базисного набору та молекулярної геометрії (наприклад, відстані між атомами). Для цієї конкретної конфігурації після вказаних перетворень результуючий кубітний Гамільтоніан є простим:
Тут представляє оператор тотожності, а — оператор Паулі-Z, що діє на один кубіт. Коефіцієнти виводяться з інтегралів, обчислених за допомогою базисного набору STO-6G при даній відстані між атомами з відповідним перетворенням.
Маючи визначений Гамільтоніан, ми тепер можемо використати VQE для обчислення енергії його основного стану. Корисно порівняти обчислену енергію основного стану з очікуваними значеннями. Для окремого, ізольованого атома водню (H) енергія основного стану точно дорівнює -0.5 Хартрі (за відсутності релятивістських ефектів). Обчислімо точну енергію основного стану нашого конкретного кубітного Гамільтоніана, визначеного вище, та порівняємо її з відповідними відомими значеннями.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Далі нам потрібна параметризована квантова схема — анзац — для підготовки пробної хвильової функції основного стану. Мета — знайти параметри , які мінімізують очікуване значення енергії . Вибір анзацу є надзвичайно важливим, оскільки він визначає набір можливих квантових станів, які може підготувати наша схема. "Хороший" анзац — той, що є достатньо гнучким для представлення стану, дуже близького до істинного основного стану досліджуваного Гамільтоніана, але не настільки складного, щоб вимагати занадто багато параметрів або занадто глибокої схеми для сучасних квантових комп'ютерів.
Тут ми спробуємо три різних однокубітних ansätze, щоб побачити, який із них забезпечує кращу "охопленість" можливих квантових станів одного кубіту. "Охопленість" означає діапазон квантових станів, які схема анзацу може продукувати, варіюючи свої параметри.
Ми використаємо три ansätze на основі різних комбінацій однокубітних обертальних гейтів:
- Анзац з одним обертальним гейтом навколо однієї осі: цей анзац використовує обертання лише навколо однієї осі (). На сфері Блоха це відповідає руху лише вздовж певного кола. Це найменш гнучкий варіант, що охоплює обмежений набір станів.
- Два ansätze з обертальними гейтами навколо двох осей: ці ansätze комбінують обертання навколо двох різних осей ( та ). Це дозволяє охопити більшу частину сфери Блоха порівняно з обертанням навколо однієї осі.
Порівнюючи результати VQE, отримані з цими трьома ansätze, ми можемо побачити, як гнучкість і охопленість простору станів анзацу впливають на нашу здатність знайти справжню енергію основного стану спрощеного Гамільтоніана. Гнучкіший анзац потенційно здатен знайти краще наближення, але може ускладнити завдання для класичного оптимізатора.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Тепер згенеруємо 5000 випадкових чисел для кожного параметра і побудуємо розподіл випадкових квантових станів, згенерованих трьома ansätze з цими випадковими параметрами. Ці параметри можна уявити як обертання навколо різних осей на сферичній поверхні. Щоб побачити розподіл квантових станів, ми використаємо сферу Блоха — тривимірну сферу, що відображає стан одного кубіту. Будь-яка точка на сфері представляє можливий стан кубіту, де північний і південний полюси подібні до класичних "0" і "1", але кубіт може перебувати в будь-якій проміжній точці, демонструючи особливі квантові властивості, як-от суперпозиція. Спочатку підготуємо необхідні функції для побудови тривимірної сфери Блоха та згенеруємо 5000 випадкових параметрів.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Подивімося, як працює наш перший анзац.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
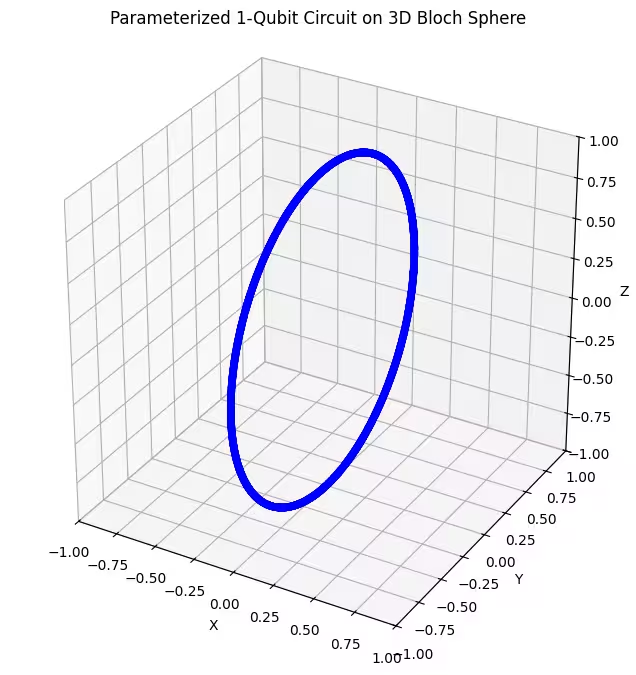
Ми бачимо, що перший анзац генерує квантові стани, розподілені у вигляді кільця на сфері Блоха. Це логічно, адже анзацу задано лише один параметр обертання. Тому він може генерувати лише стани, що обертаються навколо однієї осі. Починаючи з точки і обертаючись навколо однієї осі, завжди отримуємо кільце. Тепер перевіримо наш другий анзац, який має два ортогональних обертальних гейти — Rx і Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
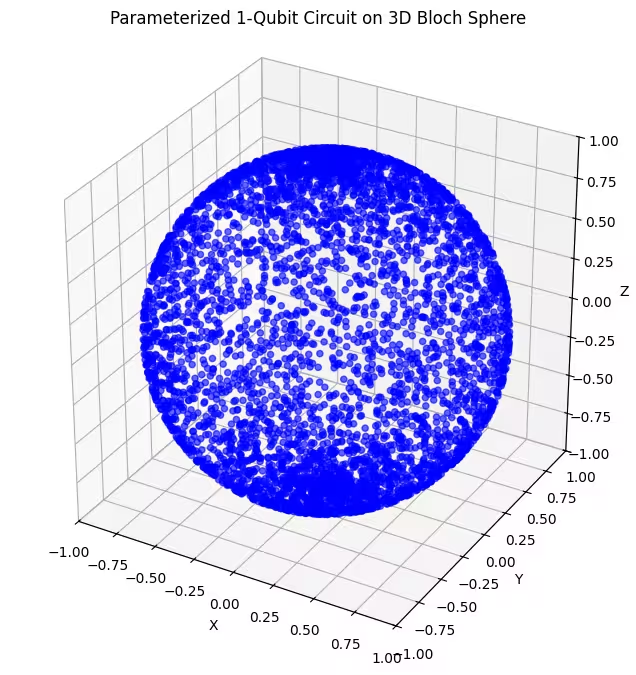
Тут видно, що наш другий анзац охоплює більшу частину сфери Блоха — але зверни увагу: точки більш сконцентровані навколо полюсів і більш розосереджені навколо екватора. Тепер час перевірити наш останній анзац.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
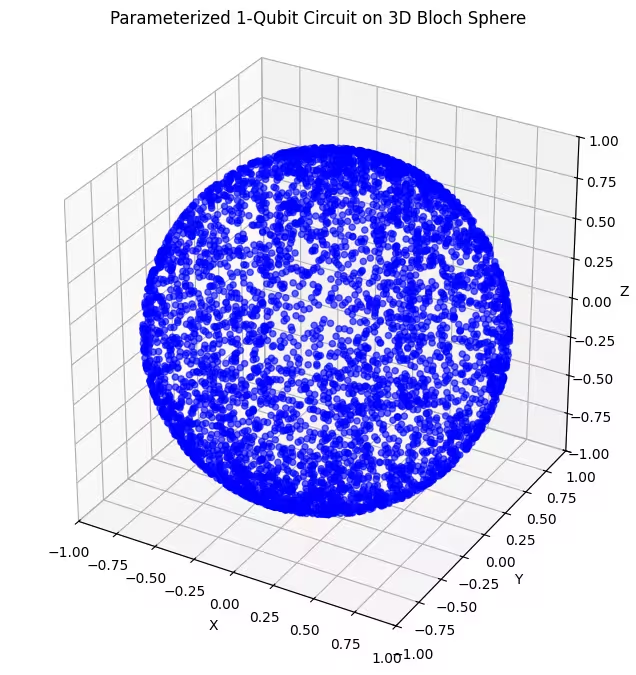
Тут видно рівномірніший розподіл квантових станів, згенерованих нашим останнім анзацом.
Як уже зазначалося, найкраще — мати знання про основний стан, який ти шукаєш, і вибирати анзац, що добре підходить для дослідження станів, близьких до нього. Наприклад, якщо ми знаємо, що наш основний стан знаходиться поблизу полюса, можна вибрати анзац 2. Для простоти ми зупинимося на анзаці 3, який рівномірно досліджує всю сферу Блоха.
Тепер, коли ми вибрали анзац, намалюємо схему.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Крок 2: Оптимізація під цільове залізо
Коли ти запускаєш обчислення на реальному квантовому комп'ютері, важливо не лише те, яку логіку реалізує квантова схема. Важливо також, які операції підтримує конкретний квантовий комп'ютер і де саме на ньому розміщені кубіти, що ти використовуєш. Чи розташовані вони поруч? Чи далеко один від одного? Тому наступний крок — переписати схему з використанням гейтів, природних для обраного квантового комп'ютера, з урахуванням розміщення кубітів. Це робиться за допомогою transpilation — після цього процесу ти побачиш, як наш простий анзац перетворюється на інший набір гейтів, а абстрактні кубіти відображаються у фізичні кубіти реального квантового комп'ютера.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Ти можеш бачити, що гейти rx, rz нашого анзацу були перетворені на послідовність гейтів rz, sx — нативних гейтів нашого бекенду. Крім того, кубіт q0 тепер відображений на п'ятий фізичний кубіт. Нам також потрібно відповідним чином перетворити Гамільтоніан:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Крок 3: Виконання на цільовому залізі
Час запустити наш VQE на реальному QPU. Для цього спочатку нам потрібна функція витрат для процесу оптимізації, яка обчислює очікуване значення Гамільтоніана для квантового стану, підготовленого анзацом. Не хвилюйся! Тобі не потрібно писати все самостійно. Ми підготували функцію для цього — просто запусти комірку нижче.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Далі ми готуємо початкові параметри для анзацу та процесу оптимізації. Можна просто використати нулі або випадкові значення. Нижче ми вибрали конкретні початкові параметри, але якщо хочеш — закоментуй або розкоментуй рядки в комірці, щоб вибрати параметри випадково з рівномірного розподілу від 0 до .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Вітаємо! Ти щойно успішно завершив свій перший квантово-хімічний експеримент. Ми бачимо невелику різницю між точною енергією основного стану Гамільтоніана та нашим результатом, але завдяки стандартній техніці пом'якшення помилок (яка виправляє помилки зчитування) ця різниця незначна. Чудовий початок!
Примітка: Кращий результат можна отримати, встановивши рівень пом'якшення помилок через resilience_level. Стандартне значення — 1, і якщо встановити вище, буде витрачено більше часу QPU, але результат може бути точнішим.
Крок 4: Постобробка
Час подивитись, як спрацював наш класичний оптимізатор. Запусти комірку нижче і поглянь на картину збіжності.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Ми розпочали з досить вдалого початкового значення, тому вже за 10 кроків отримали хороший результат. Ти можеш бачити великі та малі піки — це типова поведінка оптимізатора COBYLA: він досліджує простір, ніби не бачить ландшафту, і коригує розмір кроку після кожного вимірювання.
Перевір своє розуміння
Що ти помітив? Яку частину описаного процесу можна покращити, щоб отримати результати, ближчі до теоретичних значень або до точної енергії основного стану Гамільтоніана? Що варто врахувати?
Answer
Перше, що варто розглянути, — це зміна набору базисів, що використовуються для обчислення Гамільтоніана молекул. Як згадувалося раніше, енергія основного стану атома H дорівнює -0,5 Хартрі, і базис STO-6G, який ми обрали, недостатній для точного відтворення цього значення.
Вибір складнішого базису збільшує кількість кубітів, необхідних для Гамільтоніана; тому потрібно обирати складніший і більш підходящий анзац для хімічних задач.
Наступне, що потрібно оптимізувати, — управління шумами в QPU. Досконаліші техніки пом'якшення помилок дають кращі результати, але можуть потребувати більше часу. Також врахуй, як параметр shot_number впливає на результати.
Нарешті, кращої збіжності можна досягти, спробувавши різні оптимізатори.
Обчислення енергії основного стану молекули водню за допомогою VQE
Тепер, коли ми розглянули загальний процес VQE на прикладі атомів , обчислимо енергію основного стану молекули швидше.
Крок 1: Відображення задачі на квантові схеми та оператори
Тут ми також надаємо однокубітний Гамільтоніан, що використовує базис STO-6G і перетворення Йордана-Вігнера зі зменшенням кількості кубітів завдяки симетрії Гамільтоніана. Зауважимо, що ми використали міжатомну відстань між двома атомами водню 0.735 .
На відміну від обчислення одиночного атома водню (), для обчислення основного стану молекули водню () нам потрібно також враховувати відштовхувальну силу між ядрами двох атомів водню, крім енергії, пов'язаної з електронними орбіталями. На цьому кроці ми задамо це значення як константу, а фактичне обчислення розглянемо в перевірочній задачі.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Крок 2: Оптимізація під цільове залізо
Оскільки кількість кубітів попереднього VQE та Гамільтоніана збігається з бекендом, що використовуватиметься для виконання, ми скористаємося наявним анзацом та його оптимізованою формою.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Крок 3: Виконання на цільовому залізі
Час виконати обчислення на реальному QPU. Майже все залишається таким самим, але ми підберемо відповідну початкову точку під цей Гамільтоніан. Крім того, на ітераційному етапі деякі налаштування Estimator — примітиву для обчислення очікуваного значення Гамільтоніана для анзацу на QPU — будуть дещо відрізнятися від попередніх обчислень. Ми детальніше обговоримо цю зміну в перевірочному питанні.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Хоча VQE теоретично дає верхню межу для істинної енергії основного стану, практичні реалізації на реальному або шумному симульованому квантовому залізі, а також апроксимації при підготовці Гамільтоніана (як-от вибір базисних наборів або зменшення кількості кубітів) можуть вносити похибки, що іноді призводять до виміряної енергії, дещо нижчої за точне теоретичне значення або певний числовий орієнтир. Хоча певні похибки є, результати виглядають задовільно — особливо з огляду на невелику кількість кроків. Тепер завершимо цей VQE-розрахунок, поглянувши на роботу оптимізатора.
Крок 4: Постобробка
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Перевір своє розуміння
Обчислімо енергію ядерного відштовхування молекули , яку ми задали як константу (0,71997 Хартрі).

Скористайся законом Кулона і атомними одиницями, щоб отримати значення в Хартрі.
Answer
Оскільки обидва ядра водню мають позитивний заряд, вони відштовхуються одне від одного внаслідок електростатичної сили. Це відштовхування описується законом Кулона:
де — заряд протона, — електрична стала вакууму, а — відстань між двома ядрами, виміряна в метрах або радіусах Бора у джоулях (Дж).
Щоб обчислити цю енергію в Хартрі, потрібно перевести наведене рівняння у систему атомних одиниць (AU). В AU , , а радіус Бора () дорівнює 1 і є базовою одиницею довжини. З цими спрощеннями закон Кулона набуває вигляду:
де вимірюється в радіусах Бора ().
Щоб перевести задану відстань між ядрами з у , використовуємо таке співвідношення:
тоді стає .
Отже, енергія ядерного відштовхування для даної молекули дорівнює
Обчислення енергії реакції
Тепер скористаємось тим, що ми отримали! Ти використав VQE — варіаційний квантовий розв'язувач власних значень — для обчислення енергій основного стану атома та молекули . Залишилось використати отримані значення для розрахунку енергії реакції процесу .
Енергія реакції — це зміна енергії, що відбувається, коли речовини реагують, утворюючи нові сполуки. Уяви, що ти щось будуєш: іноді потрібно докласти енергію (як складати кубики), а іноді енергія вивільняється (як м'яч, що котиться з гори). У хімії реакції або поглинають енергію (ендотермічні), або вивільняють її (екзотермічні).
Енергія реакції процесу обчислюється за такою формулою:
Запустивши комірку нижче, ми побачимо це наочно. Тут ми використаємо точні значення енергії основного стану кожного Гамільтоніана та порівняємо енергію реакції для точного розв'язку та результатів VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Як видно з рисунку, попри деякі похибки, точна енергія основного стану Гамільтоніанів та енергія реакції, обчислена за результатами VQE, є близькими між собою — приблизно -0,2 Хартрі.
Варто зазначити, що енергія реакції цього процесу є від'ємною, а отже, у ході реакції вивільняється енергія, і утворена молекула має нижчу енергію, ніж два окремих атоми. 6. Висновок
Підсумуємо все, що ми вивчили.
Спочатку ми розглянули дві важливі техніки апроксимації, необхідні для розв'язання задач квантової хімії: варіаційний принцип і вибір базисних наборів — обидва є фундаментальними для VQE. Ми дослідили варіаційний принцип вручну, обчислюючи енергію основного стану гармонічного осцилятора.
Далі ми вивчили VQE — широко використовуваний алгоритм для обчислення енергії основного стану квантової системи. Ми запускали код для обчислення енергій основного стану атомарного водню () та молекули водню (). Зокрема, ми дізналися, що для роботи алгоритму необхідно отримати відповідний молекулярний Гамільтоніан системи та перетворити його у форму, придатну до виконання на квантовому комп'ютері. Ми також побачили, що анзац — параметризована квантова схема — потрібний для підготовки пробних квантових станів у VQE, та обговорили важливість вибору відповідної структури схеми анзацу. Крім того, ми дізналися, що VQE покладається на ітеративний процес оптимізації за допомогою класичного комп'ютера, що скеровує квантову схему до пошуку стану з найнижчою енергією, і спостерігали, як цей процес збігається.
Нарешті, ми використали обчислені через VQE енергії основного стану та , щоб розрахувати енергію реакції .
VQE — це потужний квантовий алгоритм для поточного покоління пристроїв, проте важливо усвідомлювати його обмеження. Ефективність VQE значною мірою залежить від вибору анзацу — знайти ефективно підготовлюваний анзац, здатний точно представляти справжній основний стан, стає дедалі складніше для більших і складніших молекул. Крім того, сучасне квантове залізо схильне до шумів, що може впливати на точність результатів VQE, особливо для глибших схем або більшої кількості кубітів. Попри ці виклики, VQE є фундаментальним алгоритмом, і поточні дослідження вивчають досконаліші варіаційні методи та техніки пом'якшення помилок, щоб розширити можливості квантової хімії на пристроях наступного покоління. Наприклад, розробляються алгоритми на кшталт Sample-based Quantum Diagonalization (SQD), які поєднують вибірки, отримані з квантових схем, із класичною діагоналізацією в підпросторі для покращення оцінки енергії та усунення деяких обмежень VQE — зокрема щодо ефективності вимірювань і стійкості до шумів.
Огляд і запитання
Ключові концепції:
- Варіаційний квантовий алгоритм — це обчислювальна парадигма, в якій класичний і квантовий комп'ютери працюють разом для розв'язання задачі.
- У VQE ми починаємо з Гамільтоніана нашої системи та відображаємо його на кубіти для виконання на квантовому комп'ютері. Ми обираємо параметризовану квантову схему — анзац — і виконуємо багаторазові вимірювання, варіюючи параметри анзацу, доки не буде досягнуто мінімального значення енергії. Пошук у просторі параметрів виконується за допомогою класичного оптимізатора. Щоб отримати хороші результати, необхідно обрати вдалий анзац і відповідний оптимізатор.
- Енергія реакції — це загальна зміна енергії в хімічній реакції, що визначається різницею між енергією реагентів і продуктів.
Правда/Неправда
- Варіаційний принцип стверджує, що очікуване значення енергії для будь-якої пробної хвильової функції завжди більше або рівне справжній енергії основного стану.
- Базисний набір — це сукупність функцій, що використовуються для апроксимації квантових хвильових функцій.
- VQE — це квантовий алгоритм для точного розв'язання рівняння Шредінгера для заданого Гамільтоніана.
- У VQE параметризована квантова схема (анзац) використовується для підготовки пробних хвильових функцій.
- Вибір оптимізатора у VQE (наприклад, COBYLA, SPSA або ADAM) не впливає на якість результату.
Estimatorу Qiskit використовується для безпосереднього обчислення очікуваних значень Гамільтоніанів у VQE.
Питання з вибором відповіді:
- Яка роль Гамільтоніана у VQE?
- A) Генерувати випадкові квантові стани
- B) Визначати енергію квантових станів
- C) Оптимізувати квантові схеми
- D) Створювати заплутаність
- Яка головна мета алгоритму VQE?
- A) Знайти енергію основного стану Гамільтоніана
- B) Створити заплутаність між кубітами
- C) Виконати пошук Гровера
- D) Зламати шифрування RSA
- Скільки квантових станів генерується в цьому ноутбуці для порівняння анзацу?
- A) 100
- B) 1000
- C) 5000
- D) 10 000
- Навіщо у VQE потрібен класичний оптимізатор?
- A) Для виконання квантових вимірювань
- B) Оновлювати параметри анзацу для мінімізації енергії
- C) Для заплутування кубітів
- D) Для генерування квантової випадковості
- Чому анзац розроблено параметризованим?
- A) Щоб уможливити підготовку квантового стану
- B) Щоб можна було досліджувати широкий простір квантових станів
- C) Щоб зменшити складність схеми
- D) Щоб безпосередньо вимірювати власні значення
- Яке з наведених тверджень найточніше описує вибір вдалого анзацу?
- A) Анзац обов'язково повинен рівномірно розподіляти стани по сфері Блоха, інакше він не спрацює.
- B) Анзац слід підбирати під свою систему, щоб він міг генерувати стани, близькі до основного стану.
- C) Анзац повинен генерувати випадкові стани за допомогою варіаційних параметрів.
- D) Кращий анзац завжди має більше варіаційних параметрів.
(Додатково) Додаток: Накладні витрати оптимізатора залежно від складності анзацу
VQE стикається з кількома добре відомими проблемами[ref 6], і наведені нижче пов'язані з тим, що ми вивчили вище.
- Проблеми вибору анзацу
Вибір правильного варіаційного анзацу є непростим завданням. Натхненні хімією анзаци (наприклад, UCCSD) забезпечують фізичну точність, але потребують глибоких квантових схем, тоді як апаратно-ефективні анзаци мають менш глибокі квантові схеми, але можуть не мати фізичної інтерпретованості. Крім того, багато анзаців вводять надмірну кількість варіаційних параметрів, які практично не покращують точність, але суттєво ускладнюють оптимізацію.
- Труднощі оптимізації
Ландшафт оптимізації VQE може мати ділянки, де градієнти загасають експоненціально (безплідні плато), що ускладнює ефективне оновлення варіаційних параметрів класичними оптимізаторами. Для вирішення цього дослідники намагалися використовувати різні типи оптимізаторів — градієнтні та безградієнтні, — але обидва підходи стикаються з труднощами. Градієнтні оптимізатори страждають від безплідних плато, а безградієнтні методи потребують великої кількості обчислень функції.
- Накладні витрати оптимізатора
Ще одна добре відома проблема — накладні витрати оптимізатора, що пов'язані з масштабом задачі. Квантові схеми, необхідні для VQE, збільшуються у глибину та складність зі зростанням розміру задачі; це, як правило, також збільшує кількість параметрів для оптимізації. Процес оптимізації стає нездійсненним зі зростанням кількості параметрів, що призводить до повільної збіжності та труднощів у пошуку оптимального розв'язку.
Тут ми розглянемо ці виклики, застосувавши VQE для молекули з двома різними типами анзацу.
(Примітка: це може зайняти більше часу QPU, тому можеш скористатися симулятором, якщо не маєш достатньо часу.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Тепер запустимо VQE з початковою точкою, що складається з одиниць, максимум 20 кроків, і порівняємо збіжність обох запусків.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Наведений вище графік чітко демонструє, що процес оптимізації анзацу з більшою кількістю змінних потребує більше часу для досягнення стабільної збіжності.
Замість того щоб покладатися на прості однокубітні квантові схеми та прямолінійний анзац, складність оптимізації зростає, коли потрібні більші квантові схеми й більш складно структуровані анзаци. Це підкреслює добре відому проблему у VQE: накладні витрати оптимізатора.
Дослідники продовжують розробляти різноманітні передові методології, що дають змогу використовувати квантові комп'ютери для розв'язання хімічних задач. Різноманітні навчальні матеріали можна знайти на IBM Quantum Learning.
Посилання
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)