Покращення оцінки енергії хімічного гамільтоніана за допомогою SQD
У цьому посібнику ми реалізуємо шаблон Qiskit, що демонструє, як виконувати постобробку зашумлених квантових вибірок для знаходження наближення основного стану хімічного гамільтоніана: молекули у рівноважному стані в базисному наборі 6-31G. Ми будемо дотримуватися підходу квантової діагоналізації на основі вибірок для обробки вибірок, отриманих із квантового ансатцу Circuit з 36 qubit (у цьому випадку схема LUCJ). Для врахування ефекту квантового шуму використовується техніка відновлення конфігурацій.
Шаблон можна описати у чотири кроки:
- Крок 1: Відображення на квантову задачу
- Генерація ансатцу для оцінки основного стану
- Крок 2: Оптимізація задачі
- Транспіляція ансатцу для Backend
- Крок 3: Виконання експериментів
- Отримання вибірок з ансатцу за допомогою примітиву
Sampler
- Отримання вибірок з ансатцу за допомогою примітиву
- Крок 4: Постобробка результатів
- Самоузгоджений цикл відновлення конфігурацій
- Постобробка повного набору бітрядкових вибірок з використанням апріорних знань про кількість частинок та середню заповненість орбіталей, обчислену на найостаннішій ітерації.
- Імовірнісне створення пакетів підвибірок із відновлених бітрядків.
- Проєктування та діагоналізація молекулярного гамільтоніана на кожному підпросторі вибірки.
- Збереження мінімальної енергії основного стану, знайденої в усіх пакетах, та оновлення середньої заповненості орбіталей.
- Самоузгоджений цикл відновлення конфігурацій
Для цього прикладу гамільтоніан взаємодіючих електронів має загальний вигляд:
/ — це ферміонні оператори народження/знищення, пов'язані з -м елементом базисного набору та спіном . та — одно- та двочастинкові електронні інтеграли.
Робочий процес SQD із самоузгодженим відновленням конфігурацій зображено на наступній діаграмі.
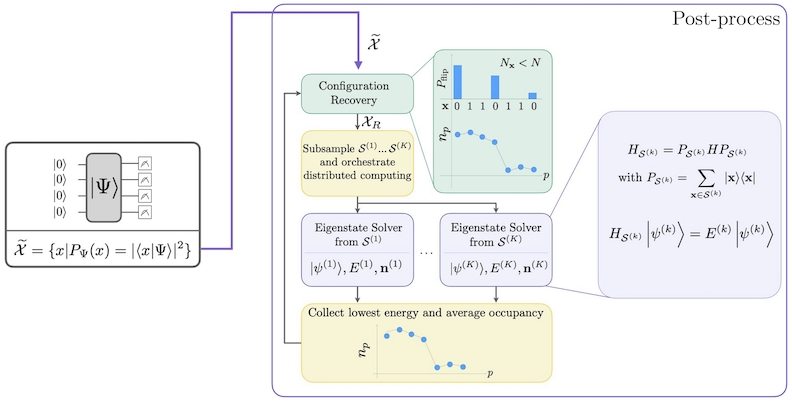
Відомо, що SQD добре працює, коли цільовий власний стан є розрідженим: хвильова функція підтримується на наборі базисних станів , розмір якого не зростає експоненціально з розміром задачі. У цьому сценарії діагоналізація гамільтоніана, спроєктованого на підпростір, визначений :
дає хороше наближення до цільового власного стану. Роль квантового пристрою полягає у виробленні вибірок елементів . Спочатку квантова схема підготовлює стан у квантовому пристрої. Використовується кодування Джордана–Вігнера. Відповідно, елементи обчислювального базису представляють стани Фока (електронні конфігурації/детермінанти). Схема вибирається у обчислювальному базисі, що дає набір зашумлених конфігурацій . Конфігурації представлені бітрядками. Набір потім передається до блоку класичної постобробки, де використовується самоузгоджена техніка відновлення конфігурацій. У рамках SQD роль квантового пристрою полягає у виробленні розподілу ймовірностей.
Крок 1: Відображення задачі на квантову схему
У цьому посібнику ми будемо наближати енергію основного стану молекули . Спочатку ми задамо молекулу та її властивості. Потім створимо ансатц локального унітарного кластерного Ястрова (LUCJ) (квантову схему) для генерації вибірок із квантового комп'ютера з метою оцінки енергії основного стану.
Спочатку задамо молекулу та її властивості.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Далі ми створимо ансатц. Ансатц LUCJ є параметризованою квантовою схемою, і ми ініціалізуємо його з амплітудами t2 та t1, отриманими з розрахунку CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Ми будемо використовувати пакет ffsim для створення та ініціалізації ансатцу з амплітудами t2 та t1, обчисленими вище. Оскільки наша молекула має замкнооболонковий стан Хартрі–Фока, ми будемо використовувати спін-збалансований варіант ансатцу UCJ, UCJOpSpinBalanced.
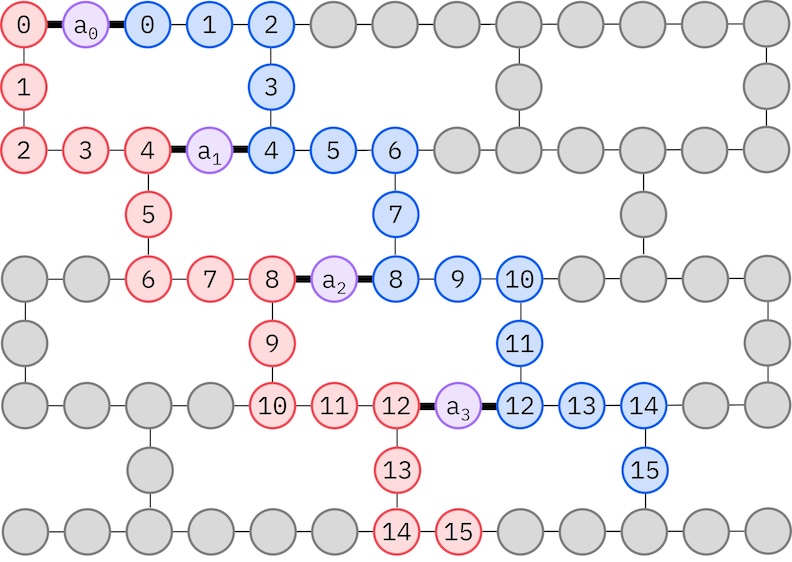
Оскільки наше цільове апаратне забезпечення IBM має топологію важкого шестикутника (heavy-hex), ми використаємо зигзагоподібний шаблон для взаємодій qubit. У цьому шаблоні орбіталі (представлені qubit) з однаковим спіном з'єднані лінійною топологією (червоні та сині кола), де кожна лінія має зигзагоподібну форму через heavy-hex зв'язність цільового апаратного забезпечення. Знову ж таки, через топологію heavy-hex, орбіталі для різних спінів мають зв'язки між кожною 4-ю орбіталлю (0, 4, 8 тощо) (фіолетові кола).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Крок 2: Оптимізація задачі
Далі ми оптимізуємо нашу схему для цільового апаратного забезпечення. Перед оптимізацією схеми нам потрібно вибрати апаратний пристрій. Ми будемо використовувати хибний 127-Qubit Backend із qiskit_ibm_runtime для емуляції реального пристрою. Щоб запустити на реальному QPU, просто заміни хибний Backend на реальний. Детальніше дивись у документації Qiskit IBM Runtime.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Далі рекомендуємо виконати такі кроки для оптимізації ансатцу та забезпечення його сумісності з апаратним забезпеченням.
- Вибери фізичні qubit (
initial_layout) із цільового апаратного забезпечення, що відповідають описаному вище зигзагоподібному шаблону. Розташування qubit за цим шаблоном призводить до ефективної, сумісної з апаратним забезпеченням схеми з меншою кількістю Gate. - Згенеруй поетапний менеджер проходів за допомогою функції generate_preset_pass_manager з Qiskit з вибраним
backendтаinitial_layout. - Встанови етап
pre_initсвого поетапного менеджера проходів наffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITвключає проходи Transpiler Qiskit, які розкладають Gate на орбітальні обертання, а потім об'єднують їх, що призводить до меншої кількості Gate у фінальній схемі. - Запусти менеджер проходів на своїй схемі.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Крок 3: Виконання експериментів
Після оптимізації схеми для виконання на апаратному забезпеченні ми готові запустити її на цільовому апаратному забезпеченні та зібрати вибірки для оцінки енергії основного стану. Оскільки у нас лише одна схема, ми будемо використовувати режим виконання завдань Qiskit Runtime і виконаємо нашу схему.
Примітка: Ми закоментували код для запуску схеми на QPU і залишили його для ознайомлення користувача. Замість запуску на реальному апаратному забезпеченні у цьому посібнику ми просто генеруємо випадкові вибірки з рівномірного розподілу.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Крок 4: Постобробка результатів
Тепер запустимо алгоритм SQD за допомогою функції diagonalize_fermionic_hamiltonian. Пояснення аргументів цієї функції дивись у документації API.
Розв'язувач, включений у доповнення SQD, використовує реалізацію вибраного CI у PySCF, зокрема pyscf.fci.selected_ci.kernel_fixed_space. Наведений нижче приклад також демонструє, як передавати іменовані аргументи до цієї функції через вбудований розв'язувач. Тут ми передаємо аргумент max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
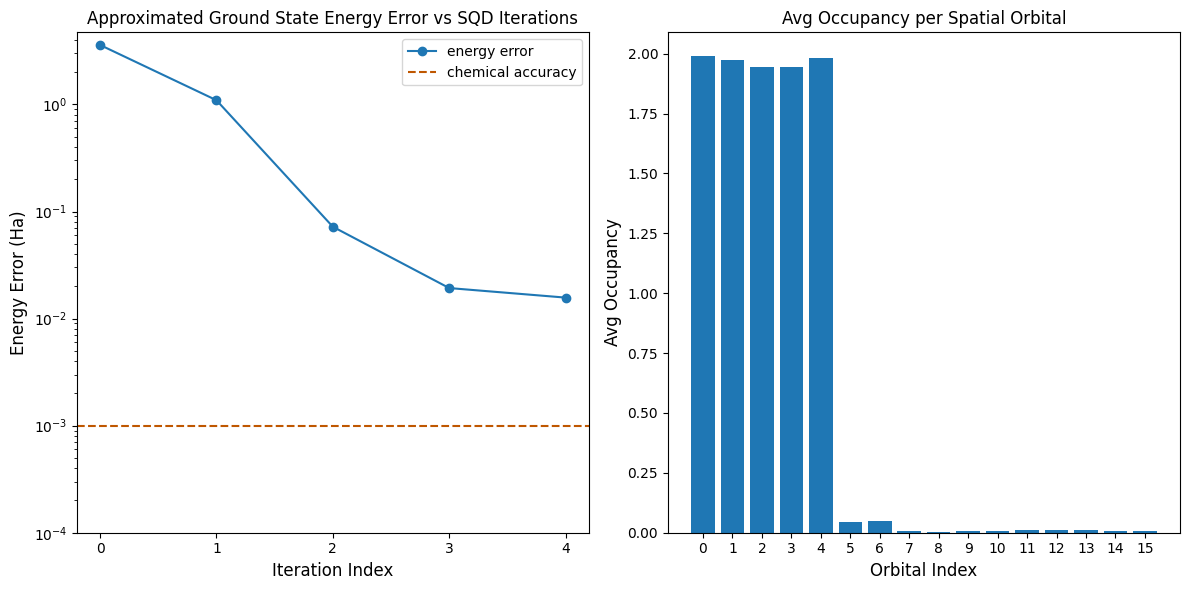
Тепер побудуємо графіки результатів.
Перший графік показує, що після кількох ітерацій ми оцінюємо енергію основного стану з точністю ~16 мГа (хімічна точність зазвичай вважається 1 ккал/моль 1.6 мГа). Пам'ятай, що квантові вибірки у цьому демо були чистим шумом. Сигнал тут походить із апріорних знань про електронну структуру та молекулярний гамільтоніан.
Другий графік показує середню заповненість кожної просторової орбіталі після фінальної ітерації. Ми бачимо, що і електрони зі спіном «вгору», і електрони зі спіном «вниз» займають перші п'ять орбіталей з високою ймовірністю у наших рішеннях.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha