Квантова діагоналізація Крилова на основі вибірок для ферміонної ґраткової моделі
Оцінка використання: дев'ять секунд на процесорі Heron r2 (ПРИМІТКА: Це лише оцінка. Твій час виконання може відрізнятися.)
Результати навчання
Після проходження цього підручника користувачі повинні розуміти:
- Як використовувати SQD Qiskit addon для апроксимації енергії основного стану ґраткової моделі за допомогою бітових рядків, відібраних із квантового процесора (QPU).
- Як використовувати ffsim для побудови схем часової еволюції для ферміонного моделювання.
- Як об'єднувати вибірки з кількох схем для постобробки за допомогою алгоритму квантової діагоналізації Крилова на основі вибірок (SKQD).
Передумови
Рекомендуємо ознайомитися з такими темами перед проходженням цього підручника:
- Квантова діагоналізація на основі вибірок для хімічного гамільтоніана
- Квантова діагоналізація Крилова для ґраткових гамільтоніанів
- Примітиви Qiskit
Передумови
Цей підручник демонструє, як використовувати квантову діагоналізацію на основі вибірок (SQD) для оцінки енергії основного стану ферміонної ґраткової моделі. Зокрема, ми досліджуємо одновимірну одноімпурітну модель Андерсона (SIAM), яка використовується для опису магнітних домішок, вбудованих у метали.
Цей підручник має подібний робочий процес до пов'язаного підручника Квантова діагоналізація на основі вибірок для хімічного гамільтоніана. Однак ключова відмінність полягає у способі побудови квантових схем. Інший підручник використовує евристичний варіаційний анзац, який є привабливим для хімічних гамільтоніанів із потенційно мільйонами членів взаємодії. З іншого боку, цей підручник використовує схеми, які апроксимують часову еволюцію за гамільтоніаном. Такі схеми можуть бути глибокими, що робить цей підхід кращим для застосування до ґраткових моделей. Вектори стану, підготовлені цими схемами, формують базис для підпростору Крилова, і завдяки цьому алгоритм доказово та ефективно збігається до основного стану за відповідних припущень.
Підхід, використаний у цьому підручнику, можна розглядати як комбінацію методів, що застосовуються в SQD та квантовій діагоналізації Крилова (KQD). Комбінований підхід іноді називають квантовою діагоналізацією Крилова на основі вибірок (SQKD). Дивись Квантова діагоналізація Крилова для ґраткових гамільтоніанів для підручника з методу KQD.
Цей підручник базується на роботі "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", до якої можна звернутися за додатковими деталями.
Одноімпурітна модель Андерсона (SIAM)
Одновимірний гамільтоніан SIAM є сумою трьох доданків:
де
Тут — це ферміонні оператори народження/знищення для вузла термостата зі спіном , — оператори народження/знищення для імпурітної моди, а . , та — дійсні числа, що описують перескок, вузлову та гібридизаційну взаємодії, а — дійсне число, що задає хімічний потенціал.
Зверни увагу, що гамільтоніан є конкретним випадком загального гамільтоніана взаємодіючих електронів,
де складається з одночастинкових доданків, які є квадратичними за ферміонними операторами народження та знищення, а складається з двочастинкових доданків, які є квартичними. Для SIAM,
а містить решту доданків гамільтоніана. Для програмного представлення гамільтоніана ми зберігаємо матрицю та тензор .
Позиційний та імпульсний базиси
Через наближену трансляційну симетрію в ми не очікуємо, що основний стан буде розрідженим у позиційному базисі (орбітальний базис, у якому гамільтоніан задано вище). Продуктивність SQD гарантується лише тоді, коли основний стан є розрідженим, тобто має значну вагу лише на невеликій кількості обчислювальних базисних станів. Для покращення розрідженості основного стану ми виконуємо моделювання в орбітальному базисі, в якому є діагональним. Ми називаємо цей базис імпульсним базисом. Оскільки є квадратичним ферміонним гамільтоніаном, його можна ефективно діагоналізувати за допомогою орбітального обертання.
Наближена часова еволюція за гамільтоніаном
Для наближення часової еволюції за гамільтоніаном ми використовуємо декомпозицію Троттера-Сузукі другого порядку,
За перетворенням Йордана-Віґнера часова еволюція за зводиться до одного вентиля CPhase між спін-вгору та спін-вниз орбіталями на вузлі домішки. Оскільки є квадратичним ферміонним гамільтоніаном, часова еволюція за зводиться до орбітального обертання.
Базисні стани Крилова , де — розмірність підпростору Крилова, формуються шляхом повторного застосування одного кроку Троттера, тому
У наступному робочому процесі на основі SQD ми будемо здійснювати вибірку з цього набору схем та постобробляти об'єднаний набір бітових рядків за допомогою SQD. Цей підхід відрізняється від того, що використовується у пов'язаному підручнику Квантова діагоналізація на основі вибірок для хімічного гамільтоніана, де вибірки отримувалися з однієї евристичної варіаційної схеми.
Вимоги
Перед початком роботи з цим підручником переконайся, що у тебе встановлено наступне:
- Qiskit SDK v1.0 або новіший, з підтримкою візуалізації
- Qiskit Runtime v0.22 або новіший (
pip install qiskit-ibm-runtime) - Додаток SQD для Qiskit v0.11 або новіший (
pip install qiskit-addon-sqd) - ffsim v0.0.72 або новіший (
pip install ffsim)
Приклад на симуляторі (малого масштабу)
Крок 1: Відображення задачі на квантову схему
Спочатку ми генеруємо гамільтоніан SIAM у позиційному базисі. Гамільтоніан представлений матрицею та тензором . Потім ми обертаємо його до імпульсного базису. У позиційному базисі ми розміщуємо домішку на першому вузлі. Однак, коли ми обертаємо до імпульсного базису, ми переміщуємо домішку до центрального вузла для полегшення взаємодій з іншими орбіталями.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
Далі ми генеруємо схеми для отримання базисних станів Крилова. Для кожного спінового різновиду початковий стан задається суперпозицією всіх можливих збуджень трьох електронів, найближчих до рівня Фермі, у 4 найближчі порожні моди, починаючи зі стану , і реалізується застосуванням семи вентилів XXPlusYYGate. Стани з часовою еволюцією отримуються послідовним застосуванням кроку Троттера другого порядку.
Для більш детального опису цієї моделі та способу проєктування схем звернись до "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Крок 2: Оптимізація задачі для квантового виконання
Далі ми оптимізуємо схему для цільового апаратного забезпечення. Наразі ми створимо загальний бекенд із заданою кількістю кубітів та набором вентилів, до яких схеми часової еволюції природно розкладаються.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Тепер ми використовуємо Qiskit для транспіляції схем на цільовий бекенд.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Крок 3: Виконання за допомогою примітивів Qiskit
Після оптимізації схем для виконання на апаратному забезпеченні ми готові запустити їх на цільовому обладнанні та зібрати зразки для оцінки енергії основного стану. Після використання примітиву Sampler для вибірки бітових рядків з кожної схеми ми об'єднуємо всі результати в один словник підрахунків та будуємо графік 20 найчастіше відібраних бітових рядків.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
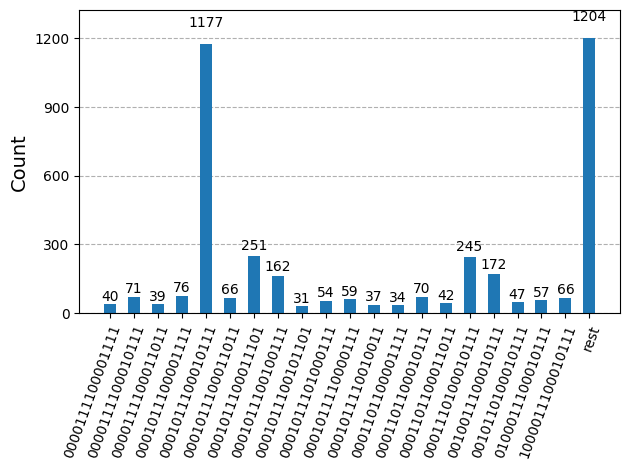
Крок 4: Постобробка та повернення результату в бажаний класичний формат
Тепер ми запускаємо алгоритм SQD за допомогою функції diagonalize_fermionic_hamiltonian. Ознайомся з документацією API для пояснення аргументів цієї функції.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
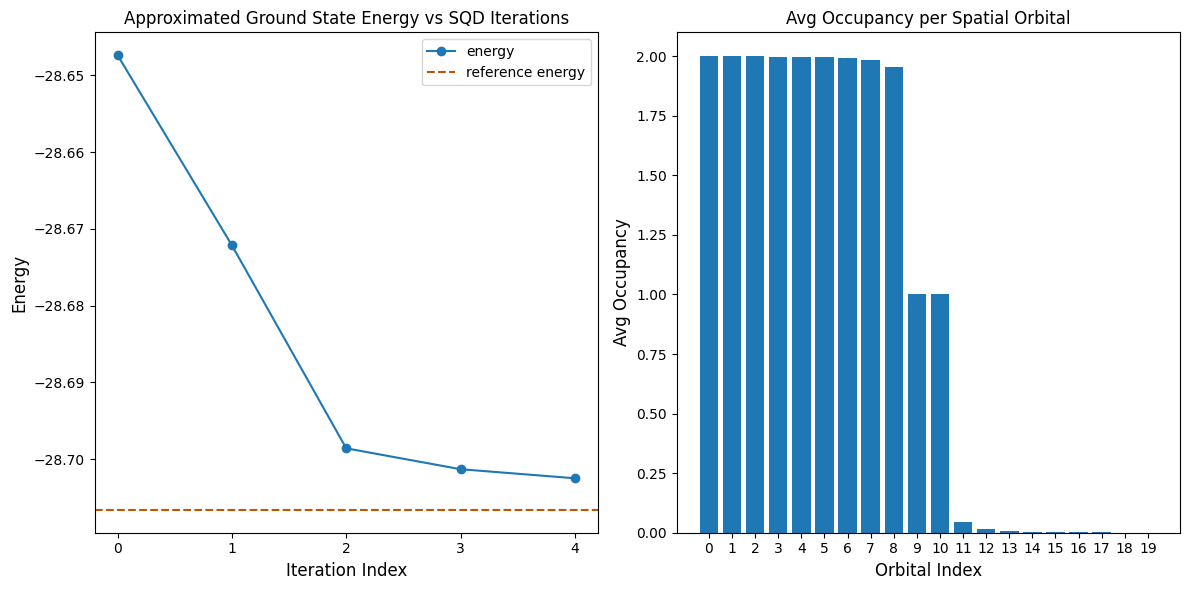
Наступна комірка коду будує графіки результатів. Перший графік показує обчислену енергію як функцію кількості ітерацій відновлення конфігурації, а другий графік показує середню заповненість кожної просторової орбіталі після фінальної ітерації. Оскільки задача настільки мала, вже перша ітерація дає результат, дуже близький до точної енергії (зверни увагу на масштаб осі y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
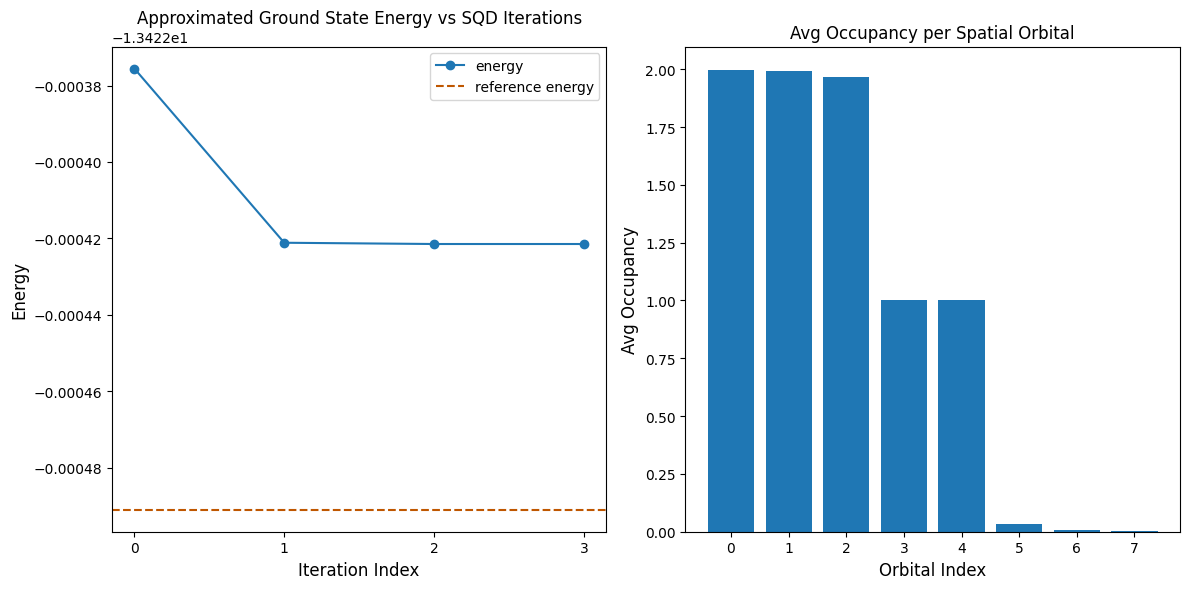
Перевір енергію
Енергія, повернута SQD, гарантовано є верхньою межею істинної енергії основного стану. Значення енергії можна перевірити, оскільки SQD також повертає коефіцієнти вектора стану, що апроксимує основний стан. Ти можеш обчислити енергію з вектора стану, використовуючи його одно- та двочастинкові зведені матриці густини, як продемонстровано в наступній комірці коду.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Великомасштабний приклад на обладнанні
Тепер ми запускаємо більш масштабний приклад на реальному QPU. Для еталонної енергії ми використовуємо результати обчислення DMRG, яке було виконано окремо.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Наступні кроки
Якщо ця робота тебе зацікавила, тебе можуть зацікавити наступні матеріали:
- Квантова діагоналізація на основі вибірок для хімічного гамільтоніана — пов'язаний підручник із використанням евристичного варіаційного анзацу замість схем Троттера
- Квантова діагоналізація Крилова для ґраткових гамільтоніанів — підручник з методу KQD
- Документація SQD addon API — довідник для функції
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization — стаття, на якій базується цей підручник