Квантова діагоналізація на основі вибірок хімічного гамільтоніана
Оцінка використання: менше однієї хвилини на процесорі Heron r2 (ПРИМІТКА: Це лише оцінка. Твій час виконання може відрізнятися.)
Результати навчання
Після проходження цього посібника користувачі повинні розуміти:
- Як використовувати SQD Qiskit addon для апроксимації енергії основного стану молекулярної системи за допомогою бітових рядків, відібраних з квантового процесора (QPU).
- Як використовувати ffsim для побудови схеми локального унітарного кластерного Яструва (LUCJ) для квантового хімічного моделювання.
Передумови
Рекомендуємо користувачам ознайомитися з такими темами перед початком роботи з цим посібником:
- Квантова хімія та друге квантування
- Використання примітива Sampler для відбору вибірок з квантових схем
Передісторія
У цьому посібнику ми показуємо, як виконати постобробку шумних квантових вибірок для апроксимації основного стану молекули азоту при рівноважній довжині зв'язку, використовуючи SQD Qiskit addon для реалізації алгоритму квантової діагоналізації на основі вибірок (SQD). Докладніше про програмне забезпечення можна дізнатися у відповідній документації, включно з простим прикладом для початку роботи.
Цей посібник рекомендований користувачам, які знайомі з квантовою хімією: зокрема, з пошуком енергій основного стану молекули. Для детального ознайомлення з робочим процесом звернися до курсу з алгоритмів квантової діагоналізації.
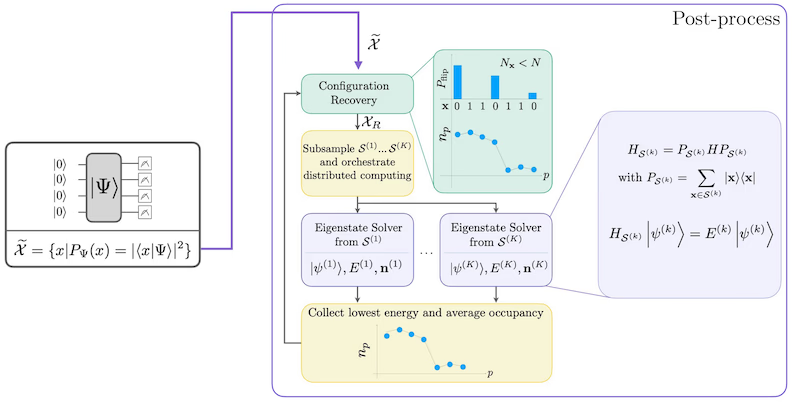
SQD — це техніка пошуку власних значень і власних векторів квантових операторів, таких як гамільтоніан квантової системи, за допомогою спільного використання квантових і розподілених класичних обчислень. Розподілені класичні обчислення використовуються для обробки вибірок, отриманих з квантового процесора, та для проектування й діагоналізації цільового гамільтоніана у підпросторі, що ними охоплюється. Робочий процес на основі SQD має такі кроки:
- Обери схему-анзац і застосуй її на квантовому комп'ютері до опорного стану (у цьому випадку — стан Хартрі-Фока).
- Відбери бітові рядки з отриманого квантового стану.
- Запусти процедуру самоузгодженого відновлення конфігурацій над бітовими рядками для отримання апроксимації основного стану.
SQD добре працює тоді, коли цільовий власний стан є розрідженим: хвильова функція підтримується на наборі базисних станів , розмір якого не зростає експоненційно зі збільшенням розміру задачі.
Квантова хімія
Гамільтоніан молекулярної системи можна записати як
де і — комплексні числа, що називаються молекулярними інтегралами і можуть бути обчислені зі специфікації молекули за допомогою комп'ютерної програми. У цьому посібнику ми обчислюємо інтеграли за допомогою програмного пакета PySCF.
Щоб дізнатися більше про те, як виводиться молекулярний гамільтоніан, звернися до підручника з квантової хімії (наприклад, Modern Quantum Chemistry Сзабо і Остлунда). Для загального пояснення того, як задачі квантової хімії відображаються на квантові комп'ютери, переглянь лекцію Mapping Problems to Qubits з Qiskit Global Summer School 2024.
Анзац локального унітарного кластерного Яструва (LUCJ)
SQD потребує квантову схему-анзац для відбору вибірок. У цьому посібнику ми використаємо анзац локального унітарного кластерного Яструва (LUCJ) завдяки поєднанню фізичної обґрунтованості та апаратної дружності. Для побудови схеми анзацу ми використаємо ffsim.
Анзац LUCJ адаптується до QPU з обмеженою зв'язністю кубітів. Спінові орбіталі відображаються на кубіти таким чином, що анзац не потребує маршрутизації зі SWAP-воротами. Обладнання IBM® має топологію кубітів типу важкого гексагону, і в цьому випадку ми можемо використати зиґзаґоподібний шаблон, зображений нижче. У цьому шаблоні орбіталі з однаковим спіном відображаються на кубіти з лінійною топологією (червоні та сині кола), а зв'язок між орбіталями різного спіну присутній кожні 4 просторові орбіталі, де зв'язок забезпечується допоміжним кубітом (фіолетові кола).
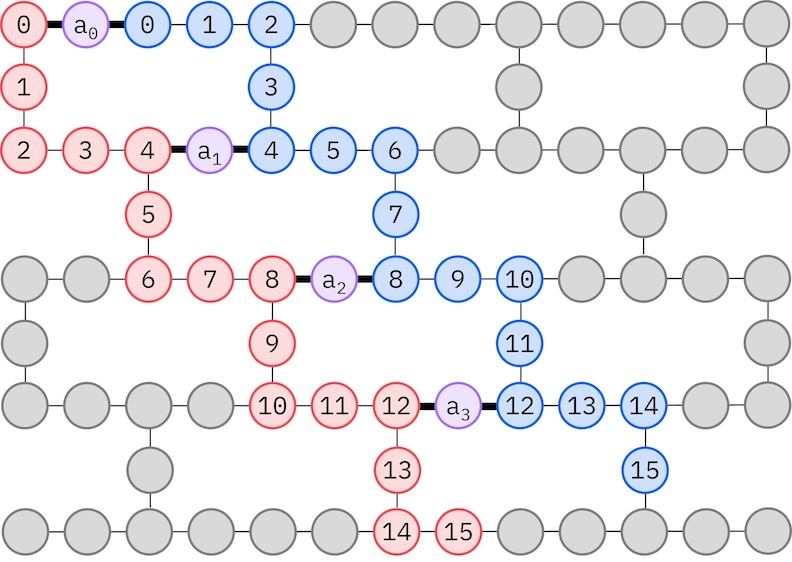
Самоузгоджене відновлення конфігурацій
Процедура самоузгодженого відновлення конфігурацій призначена для вилучення якомога більшого сигналу з шумних квантових вибірок. Оскільки молекулярний гамільтоніан зберігає число частинок і спін Z, доцільно обрати схему-анзац, яка також зберігає ці симетрії. При застосуванні до стану Хартрі-Фока, отриманий стан матиме фіксоване число частинок і спін Z у безшумних умовах. Тому половини зі спіном- і спіном- будь-якого бітового рядка, відібраного з цього стану, повинні мати таку саму вагу Геммінга, як і в стані Хартрі-Фока. Через присутність шуму в сучасних квантових процесорах деякі виміряні бітові рядки порушуватимуть цю властивість. Проста форма постселекції відкидала б такі бітові рядки, але це марнотратно, адже вони можуть ще містити певний сигнал. Процедура самоузгодженого відновлення намагається відновити частину цього сигналу при постобробці. Процедура є ітеративною і потребує на вході оцінку середньої заповненості кожної орбіталі в основному стані, яка спочатку обчислюється з необроблених вибірок. Процедура виконується в циклі, і кожна ітерація має такі кроки:
- Для кожного бітового рядка, що порушує задані симетрії, перевертай його біти за допомогою імовірнісної процедури, призначеної для наближення бітового рядка до поточної оцінки середньої орбітальної заповненості, щоб отримати новий бітовий рядок.
- Зібери всі старі та нові бітові рядки, що відповідають симетріям, і відбери підмножини фіксованого розміру, обраного заздалегідь.
- Для кожної підмножини бітових рядків проектуй гамільтоніан у підпростір, що охоплюється відповідними базисними векторами (опис цих базисних векторів дивись у попередньому розділі), і обчисли оцінку основного стану спроектованого гамільтоніана на класичному комп'ютері.
- Оновлюй оцінку середньої орбітальної заповненості за допомогою оцінки основного стану з найнижчою енергією.
Діаграма робочого процесу SQD
Робочий процес SQD зображено на такій діаграмі:
Вимоги
Перед початком цього посібника переконайся, що у тебе встановлено таке:
- Qiskit SDK v1.0 або новіший, з підтримкою візуалізації
- Qiskit Runtime v0.22 або новіший (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 або новіший (
pip install qiskit-addon-sqd) - ffsim v0.0.75 або новіший (
pip install ffsim)
Налаштування
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Маломасштабний приклад на симуляторі
У цьому посібнику ми знайдемо апроксимацію основного стану молекули азоту поблизу її рівноважної відстані зв'язку. Спочатку ми використаємо малий базисний набір STO-6G, щоб змоделювати експеримент і переконатися, що він працює.
Крок 1: Відображення класичних вхідних даних на квантову задачу
Спочатку вкажемо молекулу та її властивості.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Перед побудовою схеми анзацу LUCJ ми спочатку виконуємо розрахунок CCSD у такій комірці коду. Амплітуди і з цього розрахунку будуть використані для ініціалізації параметрів анзацу.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Тепер ми використовуємо ffsim для створення схеми анзацу. Оскільки наша молекула має замкнутий стан Хартрі-Фока, ми використовуємо спін-збалансований варіант анзацу UCJ, UCJOpSpinBalanced. Ми встановлюємо optimize=True у методі from_t_amplitudes, щоб увімкнути «стиснуте» подвійне факторизування амплітуд (дивись The local unitary cluster Jastrow (LUCJ) ansatz у документації ffsim для деталей).
Оскільки анзац LUCJ адаптується до доступної зв'язності QPU, ми маємо ініціалізувати бекенд QPU перед створенням анзацу. Зараз ми створимо загальний бекенд із картою зв'язності важкого гексагону і набором воріт, до якого анзац LUCJ природно розкладається. Потім ми використаємо ffsim.qiskit.generate_lucj_pass_manager для створення менеджера проходів, що спеціалізується на транспіляції анзацу LUCJ до заданого бекенду згідно з зиґзаґоподібним компонуванням, описаним у розділі про анзац LUCJ. Ця функція використовує евристику оцінки для мінімізації помилок, пов'язаних з обраним компонуванням, що важливо, якщо твій бекенд є реальним QPU або симулятором з моделлю шуму. Крім повернення менеджера проходів, ця функція також повертає пари зв'язку альфа-бета, які можна реалізувати на обладнанні. Якщо реалізувати не всі пари, вона видасть попередження.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Крок 2: Оптимізація для виконання на квантовому обладнанні
Далі ми оптимізуємо схему для цільового обладнання. Зазвичай цей крок передбачає ініціалізацію бекенду обладнання та менеджера проходів для нього. Однак, оскільки анзац LUCJ адаптований до зв'язності обладнання, ми вже виконали ці дії на попередньому кроці. Залишається лише запустити менеджер проходів на схемі для транспіляції її в ISA-схему, яку можна безпосередньо виконати на QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Крок 3: Виконання за допомогою примітивів Qiskit
Після оптимізації схеми для виконання на обладнанні ми готові запустити її на цільовому обладнанні та зібрати вибірки для оцінки енергії основного стану. Оскільки у нас лише одна схема, ми будемо використовувати режим виконання Job Qiskit Runtime і виконаємо нашу схему.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Крок 4: Постобробка та повернення результату у бажаному класичному форматі
Корисною метрикою для оцінки якості виводу QPU є кількість повернутих дійсних конфігурацій. Дійсна конфігурація має правильне число частинок і спін Z, тобто права половина бітового рядка має вагу Геммінга, рівну кількості електронів зі спіном вгору, а ліва половина — рівну кількості електронів зі спіном вниз. У такій комірці обчислюється частка відібраних конфігурацій, що є дійсними.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Усі бітові рядки є дійсними, оскільки ми відбираємо вибірки з схеми на безшумному симуляторі. При запуску на шумному QPU частка буде меншою за одиницю, але, сподіваємося, вона буде більшою, ніж очікувана частка при рівномірно випадковому відборі бітових рядків, яка обчислюється в такій комірці.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Тепер ми оцінюємо енергію основного стану гамільтоніана за допомогою функції diagonalize_fermionic_hamiltonian. Ця функція виконує процедуру самоузгодженого відновлення конфігурацій для ітеративного уточнення шумних квантових вибірок з метою покращення оцінки енергії. Ми передаємо функцію зворотного виклику, щоб мати змогу зберігати проміжні результати для подальшого аналізу. Пояснення аргументів функції diagonalize_fermionic_hamiltonian дивись у документації API.
Тут ми використовуємо аргумент initial_occupancies функції diagonalize_fermionic_hamiltonian, щоб вказати конфігурацію Хартрі-Фока як початкове наближення для орбітальної заповненості в основному стані. Цей підхід є доцільним для систем, де основний стан значно підтримується на конфігурації Хартрі-Фока, але він може бути неприйнятним в інших ситуаціях, хоча в таких випадках більш просунуті обчислювальні методи можуть давати кращі початкові наближення. Вказівка initial_occupancies також дозволяє запускати відновлення конфігурацій навіть якщо жодних дійсних конфігурацій не було відібрано, що може статися при відборі великої схеми на шумному QPU. Без цього аргументу відновлення конфігурацій завершиться помилкою, якщо не буде надано жодних дійсних конфігурацій.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
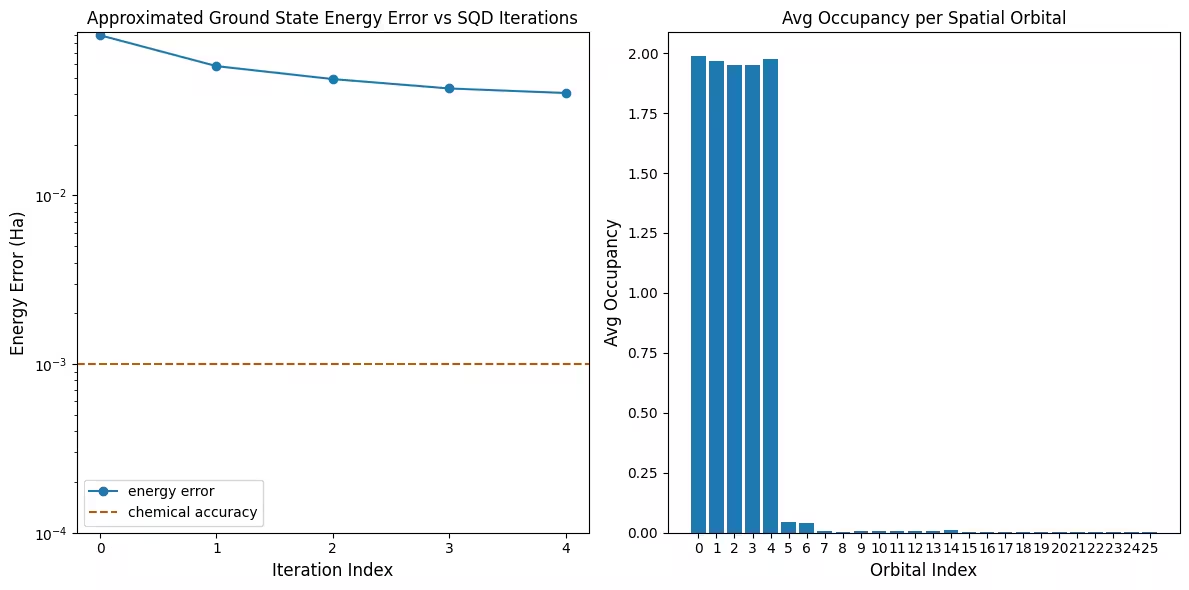
Візуалізація результатів
Перший графік показує, що в цій симуляції ми вже перебуваємо в межах 1 мГа від точної відповіді після першої ітерації (хімічна точність зазвичай приймається як 1 ккал/моль 1.6 мГа). Це мала система, і оскільки вибірки є безшумними, відновлення конфігурацій не потрібне. На більшій системі, запущеній на шумному QPU, може знадобитися кілька ітерацій відновлення конфігурацій, і остаточна точність може бути гіршою. Загалом, енергію можна покращити, дозволяючи більше ітерацій відновлення конфігурацій або збільшуючи кількість вибірок на пакет.
Другий графік показує середню заповненість кожної просторової орбіталі після фінальної ітерації. Ми бачимо, що як електрони зі спіном вгору, так і зі спіном вниз займають перші п'ять орбіталей з великою ймовірністю у наших розв'язках.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Великомасштабний приклад на обладнанні
Тепер ми запускаємо більший приклад на реальному квантовому обладнанні. Тут ми виведемо активний простір для молекули азоту з базисного набору cc-pVDZ.
Кроки 1–4
Тут ми об'єднуємо всі кроки в єдиний робочий процес у більшому масштабі, який потім запускається на реальному квантовому обладнанні.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Наступні кроки
Якщо ця робота здалася тобі цікавою, можливо, тебе зацікавлять такі матеріали:
- Sample-based Krylov quantum diagonalization of a fermionic lattice model — пов'язаний посібник, що використовує схеми часової еволюції замість варіаційного анзацу
- Scale SQD chemistry workflows with Dice solver — сторінка, що показує, як використовувати ефективніше програмне забезпечення Dice для діагоналізації
- SQD addon API documentation — довідник функції
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer — стаття, на якій заснований цей посібник