Варіаційний квантовий власний розв'язувач (VQE)
У цьому уроці ми познайомимося з варіаційним квантовим власним розв'язувачем, пояснимо його значення як фундаментального алгоритму в квантових обчисленнях, а також розглянемо його сильні та слабкі сторони. VQE сам по собі, без допоміжних методів, навряд чи достатній для сучасних квантових обчислень у промисловому масштабі. Проте він важливий як архетиповий гібридний класично-квантовий метод і є важливою основою, на якій побудовано багато більш просунутих алгоритмів.
Це відео дає огляд VQE та факторів, що впливають на його ефективність. Текст нижче доповнює деталями та реалізує VQE за допомогою Qiskit.
1. Що таке VQE?
Варіаційний квантовий власний розв'язувач — це алгоритм, що спільно використовує класичні та квантові обчислення для вирішення задачі. Існує 4 основних компоненти розрахунку VQE:
- Оператор: Зазвичай це гамільтоніан, який ми назвемо , що описує властивість системи, яку ти хочеш оптимізувати. Іншими словами, ти шукаєш власний вектор цього оператора, що відповідає мінімальному власному значенню. Ми часто називаємо цей власний вектор «основним станом».
- «Анзац» (німецьке слово, що означає «підхід»): це квантова схема, яка готує квантовий стан, що наближається до шуканого власного вектора. По суті, анзац — це сімейство квантових схем, оскільки деякі вентилі в анзаці параметризовані, тобто їм передається параметр, який ми можемо змінювати. Це сімейство квантових схем може готувати сімейство квантових станів, що наближаються до основного стану.
- Естиматор: засіб оцінювання математичного сподівання оператора у поточному варіаційному квантовому стані. Іноді нас цікавить саме це математичне сподівання, яке ми називаємо цільовою функцією. Іноді нас цікавить складніша функція, яку все одно можна виразити через одне або кілька математичних сподівань.
- Класичний оптимізатор: алгоритм, що змінює параметри, намагаючись мінімізувати цільову функцію.
Розгляньмо кожен із цих компонентів детальніше.
1.1 Оператор (гамільтоніан)
В основі задачі VQE лежить оператор, що описує систему, яка нас цікавить. Ми припускатимемо, що найменше власне значення та відповідний власний вектор цього оператора корисні для якоїсь наукової або практичної мети. Прикладами можуть бути хімічний гамільтоніан, що описує молекулу, де найменше власне значення оператора відповідає енергії основного стану молекули, а відповідний власний стан описує її геометрію або електронну конфігурацію. Або оператор може описувати вартість певного процесу, що підлягає оптимізації, а власні стани — маршрути чи практики. У деяких галузях, як-от фізика, «гамільтоніан» майже завжди означає оператор, що описує енергію фізичної системи. Але в квантових обчисленнях прийнято також називати «гамільтоніаном» квантові оператори, що описують бізнесові чи логістичні задачі. Ми будемо дотримуватися цієї конвенції.

Відображення фізичної або оптимізаційної задачі на кубіти — зазвичай нетривіальне завдання, але ці деталі не є предметом цього курсу. Загальне обговорення відображення задачі на квантовий оператор можна знайти в курсі Quantum computing in practice. Детальніший розгляд відображення хімічних задач на квантові оператори — у курсі Quantum Chemistry with VQE.
Для цілей цього курсу ми будемо вважати, що форма гамільтоніана відома. Наприклад, гамільтоніан для простої молекули водню (за певних припущень активного простору та з використанням перетворення Джордана–Вігнера) виглядає так:
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-ibm-runtime scipy
from qiskit.quantum_info import SparsePauliOp
hamiltonian = SparsePauliOp(
[
"IIII",
"IIIZ",
"IZII",
"IIZI",
"ZIII",
"IZIZ",
"IIZZ",
"ZIIZ",
"IZZI",
"ZZII",
"ZIZI",
"YYYY",
"XXYY",
"YYXX",
"XXXX",
],
coeffs=[
-0.09820182 + 0.0j,
-0.1740751 + 0.0j,
-0.1740751 + 0.0j,
0.2242933 + 0.0j,
0.2242933 + 0.0j,
0.16891402 + 0.0j,
0.1210099 + 0.0j,
0.16631441 + 0.0j,
0.16631441 + 0.0j,
0.1210099 + 0.0j,
0.17504456 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
0.04530451 + 0.0j,
],
)
Зверни увагу, що в наведеному вище гамільтоніані є такі терми, як ZZII і YYYY, що не комутують між собою. Тобто для обчислення ZZII потрібно виміряти оператор Паулі Z на кубіті 3 (серед інших вимірювань). Але для обчислення YYYY потрібно виміряти оператор Паулі Y на тому самому кубіті 3. Між операторами Y і Z на одному кубіті існує співвідношення невизначеності; ми не можемо виміряти обидва оператори одночасно. Ми повернемося до цього питання нижче і протягом усього курсу.
Наведений вище гамільтоніан є матричним оператором розміру . Діагоналізувати оператор, щоб знайти його найменше власне значення енергії, нескладно.
import numpy as np
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues), "hartrees")
The ground state energy is -1.1459778447627311 hartrees
Класичні методи грубої сили для пошуку власних значень не масштабуються до опису енергій або геометрій дуже великих атомних систем, як-от лікарські засоби чи білки. VQE — одна з перших спроб задіяти квантові обчислення для розв'язання цієї проблеми.
У цьому уроці ми зустрінемо гамільтоніани значно більші за наведений вище. Але було б недоцільно відразу долати межі можливостей VQE, перш ніж ми познайомимося з більш просунутими інструментами, що можуть доповнювати або замінювати VQE, — про них ітиметься пізніше в цьому курсі.
1.2 Анзац
Слово «ansatz» — німецьке, що означає «підхід». Правильне множина в німецькій — «ansätze», хоча часто можна зустріти «ansatzes» або «ansatze». У контексті VQE анзац — це квантова схема, яку ти використовуєш для створення багатокубітної хвильової функції, що якнайкраще наближається до основного стану системи, що вивчається, і тим самим дає найменше математичне сподівання оператора. Ця квантова схема міститиме варіаційні параметри (часто зібрані у вектор змінних ).

Вибирається початковий набір значень варіаційних параметрів. Ми позначимо унітарну операцію анзацу на схемі як . За замовчуванням усі кубіти в квантових комп'ютерах IBM® ініціалізуються у стан . Коли схема запускається, стан кубітів буде
Якби нам потрібна була лише найменша енергія (у термінах фізичних систем), ми могли б оцінити її, просто вимірюючи енергію багато разів і беручи найменше значення. Але зазвичай нас також цікавить конфігурація, що дає цю найменшу енергію або власне значення. Тому наступним кроком є оцінювання математичного сподівання гамільтоніана, яке виконується через квантові вимірювання. Це включає багато нюансів. Але якісно цей процес можна зрозуміти, зауваживши, що ймовірність виміряти енергію (знову у термінах фізичних систем) пов'язана з математичним сподіванням:
Ймовірність також пов'язана з перекриттям між власним станом і поточним станом системи :
Отже, виконуючи багато вимірювань операторів Паулі, що складають наш гамільтоніан, ми можемо оцінити математичне сподівання гамільтоніана у поточному стані системи . Наступним кроком є варіація параметрів і наближення до стану з найменшою енергією (основного стану) системи. Через наявність варіаційних параметрів анзац іноді називають варіаційною формою.
Перш ніж перейти до варіаційного процесу, зауважимо, що корисно починати зі «стану-гарного здогадування». Можливо, ти знаєш достатньо про свою систему, щоб зробити кращий початковий здогад, ніж . Наприклад, у хімічних застосуваннях прийнято ініціалізувати кубіти у стан Хартрі–Фока. Цей початковий здогад, що не містить жодних варіаційних параметрів, називається референтним станом. Позначимо квантову схему для створення референтного стану як . Коли важливо відрізнити референтний стан від решти анзацу, використовуємо: Еквівалентно
1.3 Естиматор
Нам потрібен спосіб оцінити математичне сподівання гамільтоніана у конкретному варіаційному стані . Якби ми могли безпосередньо виміряти весь оператор , це зводилося б до виконання багатьох (скажімо, ) вимірювань і усереднення виміряних значень:
Тут символ нагадує нам, що це математичне сподівання було б точним лише у границі . Але при тисячах вимірювань на схемі похибка вибірки математичного сподівання є досить малою. Є й інші міркування, як-от шум, що стають проблемою при дуже точних розрахунках.
Однак виміряти цілком, як правило, неможливо. може містити кілька некомутуючих операторів Паулі X, Y і Z. Тому гамільтоніан треба розбити на групи операторів, що можна виміряти одночасно, і кожну таку групу оцінити окремо, а потім об'єднати результати для отримання математичного сподівання. Ми повернемося до цього детальніше в наступному уроці, де розглянемо масштабування класичних і квантових підходів. Ця складність вимірювань — одна з причин, чому нам потрібен високоефективний код для такого оцінювання. У цьому та наступних уроках ми будемо використовувати для цього примітив Qiskit Runtime Estimator.
1.4 Класичні оптимізатори
Класичний оптимізатор — це будь-який класичний алгоритм, призначений для пошуку екстремумів цільової функції (як правило, мінімуму). Вони шукають в просторі можливих параметрів набір, що мінімізує функцію, яка нас цікавить. Їх можна широко розділити на методи на основі градієнта, що використовують інформацію про градієнт, і безградієнтні методи, що працюють як оптимізатори «чорного ящика». Вибір класичного оптимізатора може суттєво впливати на продуктивність алгоритму, особливо за наявності шуму на квантовому обладнанні. Популярні оптимізатори в цій галузі включають Adam, AMSGrad і SPSA, що продемонстрували обнадійливі результати у зашумлених середовищах. Більш традиційні оптимізатори включають COBYLA і SLSQP.
Типовий робочий процес (продемонстрований у розділі 3.3) — використовувати один із цих алгоритмів як метод всередині мінімізатора, наприклад функції minimize з scipy. Вона приймає такі аргументи:
- Деяку функцію для мінімізації. Це часто математичне сподівання енергії. Але їх загалом називають «цільовими функціями».
- Набір параметрів, з яких починається пошук. Часто позначається або .
- Аргументи, включно з аргументами цільової функції. У квантових обчисленнях з Qiskit ці аргументи включатимуть анзац, гамільтоніан і естиматор, про який детальніше йдеться в наступному підрозділі.
- «Метод» мінімізації. Це конкретний алгоритм пошуку в просторі параметрів. Тут ми вказуємо, наприклад, COBYLA або SLSQP.
- Опції. Доступні опції можуть різнитися залежно від методу. Але приклад, який включають практично всі методи, — максимальна кількість ітерацій оптимізатора до завершення пошуку: 'maxiter'.

На кожному ітераційному кроці математичне сподівання гамільтоніана оцінюється за допомогою багатьох вимірювань. Ця оцінена енергія повертається цільовою функцією, і мінімізатор оновлює свою інформацію про енергетичний ландшафт. Те, що саме робить оптимізатор для вибору наступного кроку, залежить від методу. Деякі використовують градієнти і обирають напрямок найкрутішого спуску. Інші можуть враховувати шум і вимагати, щоб цільова функція зменшилася на значну величину, перш ніж прийняти, що справжня енергія спадає у цьому напрямку.
# Example syntax for minimization
# from scipy.optimize import minimize
# res = minimize(cost_func, x0, args=(ansatz, hamiltonian, estimator), method="cobyla",
# options={'maxiter': 200})
1.5 Варіаційний принцип
У цьому контексті варіаційний принцип дуже важливий; він стверджує, що жодна варіаційна хвильова функція не може дати математичне сподівання енергії (або вартості) нижче, ніж те, що дає хвильова функція основного стану. Математично,
Це легко перевірити, якщо зауважити, що множина всіх власних станів оператора утворює повний базис простору Гільберта. Іншими словами, будь-який стан і зокрема можна записати як зважену (нормовану) суму цих власних станів :
де — константи, що підлягають визначенню, і . Ми залишаємо це як вправу для читача. Але зверни увагу на висновок: варіаційний стан, що дає найменше математичне сподівання енергії, і є найкращою оцінкою справжнього основного стану.
Перевір своє розуміння
Доведи математично, що для будь-якого варіаційного стану .
Answer
Використовуючи дане розкладання варіаційного стану за власними станами енергії,
можемо записати варіаційне математичне сподівання енергії як
Для всіх коефіцієнтів . Тому можемо записати
2. Порівняння з класичним робочим процесом
Припустимо, нас цікавить матриця з N рядками і N стовпцями. Нехай матриця така велика, що точна діагоналізація неможлива. Припустімо також, що ти знаєш достатньо про задачу, щоб зробити деякі здогади про загальну структуру цільового власного стану, і хочеш дослідити стани, схожі на твій початковий здогад, щоб перевірити, чи можна знизити вартість/енергію. Це варіаційний підхід, і він є одним із методів, що застосовується, коли точна діагоналізація неможлива.
2.1 Класичний робочий процес
На класичному комп'ютері це виглядало б так:
- Зроби здогад-стан з певними параметрами , які ти варіюватимеш: . Хоча цей початковий здогад може бути випадковим, це небажано. Ми хочемо якомога більше використовувати знання задачі для налаштування здогаду.
- Обчисли математичне сподівання оператора при системі у цьому стані:
- Змінюй варіаційні параметри та повторюй: .
- Використовуй накопичену інформацію про ландшафт можливих станів у варіаційному підпросторі, щоб робити дедалі кращі здогади і наближатися до цільового стану. Варіаційний принцип гарантує, що наш варіаційний стан не може дати власне значення нижче, ніж у цільового основного стану. Отже, чим менше математичне сподівання, тим краще наближення до основного стану:
Розгляньмо складність кожного кроку цього підходу. Встановлення або оновлення параметрів є обчислювально простим; складність тут полягає у виборі корисних, фізично мотивованих початкових параметрів. Використання накопиченої інформації з попередніх ітерацій для оновлення параметрів таким чином, щоб наближатися до основного стану, є нетривіальним. Але існують класичні алгоритми оптимізації, що роблять це досить ефективно. Ця класична оптимізація є дорогою лише тому, що може вимагати багатьох ітерацій; у найгіршому випадку кількість ітерацій може масштабуватися експоненційно з N. Найбільш обчислювально дорогим одиничним кроком майже напевно є обчислення математичного сподівання матриці для заданого стану :
Матриця повинна діяти на вектор з елементів, що відповідає: операціям множення у найгіршому випадку. Це потрібно виконувати при кожній ітерації параметрів. Для надзвичайно великих матриць це має значну обчислювальну вартість.
2.2 Квантовий робочий процес і комутуючі групи Паулі
Тепер уяви, що цю частину обчислення перекладаємо на квантовий комп'ютер. Замість того щоб обчислювати математичне сподівання, ти оцінюєш його, готуючи стан на квантовому комп'ютері за допомогою варіаційного анзацу, а потім роблячи вимірювання.
Це може звучати простіше, ніж є насправді. загалом нелегко виміряти. Наприклад, він може складатися з багатьох некомутуючих операторів Паулі X, Y і Z. Але можна записати як лінійну комбінацію термів , кожен з яких легко виміряти (наприклад, оператори Паулі або групи покубітно комутуючих операторів Паулі). Математичне сподівання у деякому стані є зваженою сумою математичних сподівань складових термів . Цей вираз справедливий для будь-якого стану , але ми конкретно будемо використовувати його для наших варіаційних станів .
де — рядок Паулі на кшталт IZZX…XIYX або кілька таких рядків, що комутують між собою. Тому опис математичного сподівання, що точніше відображає реалії вимірювань на квантових комп'ютерах, має вигляд
І в контексті нашої варіаційної хвильової функції:
Кожен із термів можна виміряти разів, отримуючи зразки вимірювань при , і повертає математичне сподівання та стандартне відхилення . Ми можемо підсумувати ці терми та поширити похибки через суму, отримавши загальне математичне сподівання і стандартне відхилення .
Це не вимагає великомасштабного множення і жодного процесу, що обов'язково масштабується як . Натомість це вимагає багатьох вимірювань на квантовому комп'ютері. Якщо їх потрібно не надто багато, такий підхід може бути ефективним. І це і є квантова частина VQE.
Але поговорімо про причини, чому це може бути неефективним. Одна причина великої кількості вимірювань — зменшення статистичної невизначеності у твоїх оцінках для розрахунків із дуже високою точністю. Інша причина — кількість рядків Паулі, потрібних для охоплення всієї матриці. Оскільки матриці Паулі (разом з одиничною: X, Y, Z і I) охоплюють простір усіх операторів заданої розмірності, ми гарантовано можемо записати нашу матрицю як зважену суму операторів Паулі, як ми робили раніше.
де — рядок Паулі, що діє на всіх кубітах, що описують твою систему, на кшталт IZZX…XIYX, або кілька таких рядків, що комутують між собою. Нагадаємо, що Qiskit використовує нотацію little endian, де -й оператор Паулі справа діє на -й кубіт. Тому ми можемо виміряти наш оператор, вимірюючи серію операторів Паулі.
Але ми не можемо виміряти всі ці оператори Паулі одночасно. Оператори Паулі (крім I) не комутують між собою, якщо вони пов'язані з одним і тим самим кубітом. Наприклад, ми можемо виміряти IZIZ і ZZXZ одночасно, тому що для третього кубіта можна одночасно виміряти I і Z, і для першого кубіта можна одночасно знати I і X. Але ми не можемо одночасно виміряти ZZZZ і ZZZX, тому що Z і X не комутують, і обидва діють на 0-й кубіт. Досвідчені читачі можуть пригадати, що дві групи операторів Паулі можуть комутувати як множина, навіть якщо вимірювання для окремих кубітів не комутують. Estimator використовує тензорно-добуткові вимірювання Паулі (через повороти базису), що відповідає групуванню операторів, які побітово комутують по кубітах. Тому щоб одночасно оцінити два рядки (A і B) операторів Паулі за допомогою Estimator, оператори Паулі для кожного кубіта в A і B повинні комутувати. Це означає, що ми також не можемо виміряти ZZZZ і ZZXX одночасно.

Тому ми розкладаємо матрицю у суму операторів Паулі, що діють на різних кубітах. Деякі елементи цієї суми можна виміряти всі одночасно; ми називаємо це групою комутуючих Паулі. Залежно від кількості некомутуючих термів нам може знадобитися багато таких груп. Позначимо кількість таких груп комутуючих рядків Паулі . Якщо мале, це може добре спрацювати. Якщо у мільйони груп, це буде непрактично.
Процеси, необхідні для оцінювання математичного сподівання, зібрано в примітиві Qiskit Runtime Estimator. Щоб дізнатися більше про Estimator, дивися довідник API у документації IBM Quantum®. Можна просто використовувати Estimator напряму, але Estimator повертає набагато більше, ніж просто найменше власне значення енергії. Наприклад, він також повертає інформацію про стандартну похибку ансамблю. Тому в контексті задач мінімізації Estimator часто зустрічається всередині цільової функції. Щоб дізнатися більше про вхідні та вихідні дані Estimator, дивися цей посібник у документації IBM Quantum.
Ти записуєш математичне сподівання (або цільову функцію) для набору параметрів , що використовується у твоєму стані, а потім оновлюєш параметри. З часом ти можеш використовувати оцінені значення математичних сподівань або цільової функції, щоб апроксимувати градієнт цільової функції у підпросторі станів, що досліджується анзацом. Існують як градієнтні, так і безградієнтні класичні оптимізатори. Обидва страждають від потенційних проблем навчуваності, як-от множинні локальні мінімуми та великі ділянки простору параметрів з майже нульовим градієнтом, що називаються безплідними плато.

2.3 Фактори, що визначають обчислювальну вартість
VQE не вирішить усіх твоїх найскладніших задач квантової хімії. Ні. Але бути кращим у всіх обчисленнях — не мета. Ми змістили те, що визначає обчислювальну вартість.

Ми перейшли від процесу, складність якого залежить лише від розмірності матриці, до такого, що залежить від необхідної точності та кількості некомутуючих операторів Паулі, що складають матрицю. Остання частина не має аналога в класичних обчисленнях.
На основі цих залежностей для розріджених матриць або матриць з малою кількістю некомутуючих рядків Паулі цей процес може бути корисним. Це стосується, наприклад, систем взаємодіючих спінів. Для щільних матриць це може бути менш корисним. Ми знаємо, наприклад, що хімічні системи часто мають гамільтоніани з сотнями, тисячами і навіть мільйонами рядків Паулі. Проводилася цікава робота зі скорочення кількості таких термів. Але для хімічних систем можуть краще підійти деякі інші алгоритми, що розглядаються в цьому курсі.
Перевір своє розуміння
Розглянь гамільтоніан на чотирьох кубітах, що містить терми:
IIXX, IIXZ, IIZZ, IZXZ, IXXZ, ZZXZ, XZXZ, ZIXZ, ZZZZ, XXXX
Ти хочеш відсортувати ці терми на групи так, щоб усі терми в групі можна було виміряти одночасно. Яка найменша кількість таких груп, щоб усі терми були враховані?
Answer
Це можна зробити у 4 групах. Зауважимо, що такі рішення зазвичай не є унікальними.
IIXX, XXXX, IIZZ, ZZZZ
IIXZ, IZXZ, ZIXZ, ZZXZ
IXXZ
XZXZ
Що, на твою думку, зазвичай ускладнює квантову хімію з VQE: кількість термів у гамільтоніані чи пошук гарного анзацу?
Answer
Виявляється, існують анзаці, що добре оптимізовані для хімічних контекстів. Кількість термів у гамільтоніані і, отже, кількість необхідних вимірювань зазвичай спричиняє більше проблем.
3. Приклад гамільтоніана
Застосуймо цей алгоритм на практиці, використовуючи невелику матрицю гамільтоніана, щоб побачити, що відбувається на кожному кроці. Ми будемо застосовувати фреймворк Qiskit patterns:
-Крок 1: Відображення задачі на квантові схеми та оператори -Крок 2: Оптимізація для цільового апаратного забезпечення -Крок 3: Виконання на цільовому апаратному забезпеченні -Крок 4: Постобробка результатів
3.1 Крок 1: Відображення задачі на квантові схеми та оператори
Використаємо гамільтоніан, визначений вище з хімічного контексту. Починаємо з деяких загальних імпортів.
# General imports
import numpy as np
# SciPy minimizer routine
from scipy.optimize import minimize
# Plotting functions
import matplotlib.pyplot as plt
Знову ж таки, припускаємо, що гамільтоніан відомий. Тут ми використаємо надзвичайно малий гамільтоніан, оскільки інші методи, розглянуті в цьому курсі, будуть ефективнішими для вирішення більших задач.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
hamiltonian = SparsePauliOp.from_list(
[("YZ", 0.3980), ("ZI", -0.3980), ("ZZ", -0.0113), ("XX", 0.1810)]
)
A = np.array(hamiltonian)
eigenvalues, eigenvectors = np.linalg.eigh(A)
print("The ground state energy is ", min(eigenvalues))
The ground state energy is -0.702930394459531
У Qiskit є багато готових варіантів анзацу. Ми використаємо efficient_su2.
# Pre-defined ansatz circuit and operator class for Hamiltonian
from qiskit.circuit.library import efficient_su2
# Note that it is more common to place initial 'h' gates outside the ansatz.
# Here we specifically wanted this layer structure.
ansatz = efficient_su2(
hamiltonian.num_qubits, su2_gates=["h", "rz", "y"], entanglement="circular", reps=1
)
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.decompose().draw("mpl", style="iqp")
This circuit has 4 parameters
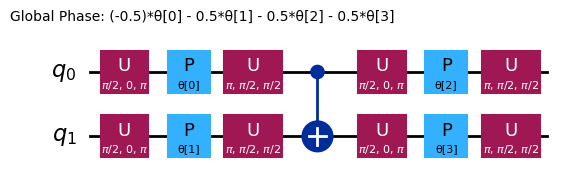
Різні анзаці матимуть різні структури заплутування та різні вентилі обертання. Показаний тут використовує вентилі CNOT для заплутування та вентилі Y і параметризовані вентилі RZ для обертань. Зверни увагу на розмір цього простору параметрів; це означає, що ми повинні мінімізувати цільову функцію за 4 змінними (параметрами для вентилів RZ). Це можна масштабувати, але не нескінченно. Запуск аналогічної задачі на 4 кубітах з параметром reps за замовчуванням 3 для efficient_su2 дає 16 варіаційних параметрів.
3.2 Крок 2: Оптимізація для цільового апаратного забезпечення
Ансац було написано з використанням знайомих вентилів, але наш контур треба транспілювати, щоб задіяти базові вентилі, доступні на конкретному квантовому комп'ютері. Вибираємо найменш завантажений бекенд.
# runtime imports
from qiskit_ibm_runtime import QiskitRuntimeService, Session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# To run on hardware, select the backend with the fewest number of jobs in the queue
service = QiskitRuntimeService()
backend = service.least_busy(operational=True, simulator=False)
print(backend)
<IBMBackend('ibm_torino')>
Тепер можемо транспілювати наш контур для цього апаратного забезпечення та візуалізувати транспілований ансац.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")

Зверни увагу: вентилі змінились, а кубіти нашого абстрактного контуру відображено на кубіти з іншими номерами на квантовому комп'ютері. Щоб результати були коректними, гамільтоніан необхідно відобразити ідентично.
hamiltonian_isa = hamiltonian.apply_layout(layout=ansatz_isa.layout)
3.3 Крок 3: Виконання на цільовому апаратному забезпеченні
3.3.1 Виведення значень
Тут ми визначаємо функцію витрат, яка приймає як аргументи структури, побудовані на попередніх кроках: параметри, ансац і гамільтоніан. Вона також використовує estimator, який ми ще не визначили. Ми включаємо код для відстеження історії функції витрат, щоб можна було перевірити поведінку збіжності.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from Estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
Дуже вигідно, якщо ти можеш обрати початкові значення параметрів, спираючись на знання задачі та характеристики цільового стану. Ми не будемо робити жодних припущень і використаємо випадкові початкові значення.
x0 = 2 * np.pi * np.random.random(num_params)
# This required 13 min, 20 s QPU time on an Eagle processor, 28 min total time.
with Session(backend=backend) as session:
estimator = Estimator(mode=session)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 50},
)
Iters. done: 1 [Current cost: 0.010575798722044727]
Iters. done: 2 [Current cost: 0.004040015974440895]
Iters. done: 3 [Current cost: 0.0020213258785942503]
Iters. done: 4 [Current cost: 0.18723082446726014]
Iters. done: 5 [Current cost: -0.2746792152068885]
Iters. done: 6 [Current cost: -0.3094547651648519]
Iters. done: 7 [Current cost: -0.05281985428356641]
Iters. done: 8 [Current cost: 0.00808560303514377]
Iters. done: 9 [Current cost: -0.0014821685303514388]
Iters. done: 10 [Current cost: -0.004759824281150161]
Iters. done: 11 [Current cost: 0.09942328705995292]
Iters. done: 12 [Current cost: 0.01092366214057508]
Iters. done: 13 [Current cost: 0.05017497496069776]
Iters. done: 14 [Current cost: 0.13028868414310696]
Iters. done: 15 [Current cost: 0.013747803514376994]
Iters. done: 16 [Current cost: 0.2583072432944498]
Iters. done: 17 [Current cost: -0.14422125655131562]
Iters. done: 18 [Current cost: -0.0004950150347678081]
Iters. done: 19 [Current cost: 0.00681082268370607]
Iters. done: 20 [Current cost: -0.0023377795527156544]
Iters. done: 21 [Current cost: 0.6027665591169237]
Iters. done: 22 [Current cost: 0.00596641373801917]
Iters. done: 23 [Current cost: -0.008318769968051117]
Iters. done: 24 [Current cost: -0.00026683306709265246]
Iters. done: 25 [Current cost: -0.007648222843450479]
Iters. done: 26 [Current cost: 0.004121086261980831]
Iters. done: 27 [Current cost: -0.004075019968051117]
Iters. done: 28 [Current cost: -0.004419369009584665]
Iters. done: 29 [Current cost: 0.213185460054037]
Iters. done: 30 [Current cost: -0.06505919572162797]
Iters. done: 31 [Current cost: -0.5334241316590271]
Iters. done: 32 [Current cost: 0.00218370607028754]
Iters. done: 33 [Current cost: 0.09579352143666908]
Iters. done: 34 [Current cost: -0.009274800319488819]
Iters. done: 35 [Current cost: -0.44395141360688106]
Iters. done: 36 [Current cost: 0.011747104632587858]
Iters. done: 37 [Current cost: -0.003344149361022364]
Iters. done: 38 [Current cost: 0.19138183916486304]
Iters. done: 39 [Current cost: 0.013513931813145209]
Можна переглянути вихідні дані безпосередньо.
res
message: Return from COBYLA because the trust region radius reaches its lower bound.
success: True
status: 0
fun: -0.5334241316590271
x: [ 1.024e+00 6.459e+00 3.625e+00 4.007e+00]
nfev: 39
maxcv: 0.0
3.4 Крок 4: Постобробка результатів
Якщо процедура завершується коректно, значення у нашому словнику мають відповідати вектору розв'язку та загальній кількості обчислень функції відповідно. Це легко перевірити:
cost_history_dict
{'prev_vector': array([1.02397956, 6.45886604, 3.62479262, 4.00744128]),
'iters': 39,
'cost_history': [np.float64(0.010575798722044727),
np.float64(0.004040015974440895),
np.float64(0.0020213258785942503),
np.float64(0.18723082446726014),
np.float64(-0.2746792152068885),
np.float64(-0.3094547651648519),
np.float64(-0.05281985428356641),
np.float64(0.00808560303514377),
np.float64(-0.0014821685303514388),
np.float64(-0.004759824281150161),
np.float64(0.09942328705995292),
np.float64(0.01092366214057508),
np.float64(0.05017497496069776),
np.float64(0.13028868414310696),
np.float64(0.013747803514376994),
np.float64(0.2583072432944498),
np.float64(-0.14422125655131562),
np.float64(-0.0004950150347678081),
np.float64(0.00681082268370607),
np.float64(-0.0023377795527156544),
np.float64(0.6027665591169237),
np.float64(0.00596641373801917),
np.float64(-0.008318769968051117),
np.float64(-0.00026683306709265246),
np.float64(-0.007648222843450479),
np.float64(0.004121086261980831),
np.float64(-0.004075019968051117),
np.float64(-0.004419369009584665),
np.float64(0.213185460054037),
np.float64(-0.06505919572162797),
np.float64(-0.5334241316590271),
np.float64(0.00218370607028754),
np.float64(0.09579352143666908),
np.float64(-0.009274800319488819),
np.float64(-0.44395141360688106),
np.float64(0.011747104632587858),
np.float64(-0.003344149361022364),
np.float64(0.19138183916486304),
np.float64(0.013513931813145209)]}
fig, ax = plt.subplots()
x = np.linspace(0, 10, 50)
# Define the constant function
constant = -0.7029
y_constant = np.full_like(x, constant)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

IBM Quantum пропонує й інші навчальні матеріали, пов'язані з VQE. Якщо ти готовий застосувати VQE на практиці, ознайомся з нашим підручником: Оцінка енергії основного стану ланцюга Гайзенберга за допомогою VQE. Якщо тобі потрібна докладніша інформація про побудову молекулярних гамільтоніанів, дивись цей урок у нашому курсі Квантова хімія з VQE. Якщо тебе цікавить глибше розуміння варіаційних алгоритмів на зразок VQE, рекомендуємо курс Variational Algorithm Design.
Перевір своє розуміння
У цьому розділі ми обчислили енергію основного стану за гамільтоніаном. Якщо б ми хотіли застосувати це, наприклад, для визначення геометрії молекули, як би ми це розширили?
Answer
Нам потрібно було б ввести змінні для міжатомних відстаней і кутів між зв'язками та варіювати їх. Для кожного варіанту цих параметрів ми отримували б новий гамільтоніан (оскільки оператори, що описують енергію, безумовно залежать від геометрії). Для кожного такого гамільтоніана, відображеного на кубіти, потрібно проводити оптимізацію, аналогічну описаній вище. З усіх тих численних збіжних задач оптимізації геометрія з найнижчою енергією і буде тією, яку обирає природа. Це значно складніше, ніж показано вище. Таке обчислення для найпростішої молекули виконується тут.
4. Зв'язок VQE з іншими методами
У цьому розділі ми розглянемо переваги та недоліки оригінального підходу VQE і вкажемо на його зв'язок з іншими, більш сучасними алгоритмами.
4.1 Сильні та слабкі сторони VQE
Деякі переваги вже були згадані. Серед них:
- Придатність до сучасного апаратного забезпечення: деякі квантові алгоритми вимагають значно нижчих рівнів помилок і наближення до великомасштабної відмовостійкості. VQE — ні; його можна реалізувати на сучасних квантових комп'ютерах.
- Мілкі контури: VQE часто використовує відносно мілкі квантові контури. Це робить VQE менш чутливим до накопичених помилок вентилів і придатним для багатьох технік мітигації помилок. Звичайно, контури не завжди мілкі — це залежить від обраного ансацу.
- Універсальність: VQE можна (в принципі) застосувати до будь-якої задачі, яку можна звести до проблеми власних значень/власних векторів. Існує багато застережень, через які VQE є непрактичним або невигідним для певних задач. Деякі з них наведені нижче.
Деякі слабкі сторони VQE та задачі, для яких він є непрактичним, також були описані вище. Серед них:
- Евристична природа: VQE не гарантує збіжності до правильної енергії основного стану, оскільки його ефективність залежить від вибору ансацу та методів оптимізації[1-2]. Якщо обрано поганий ансац, якому бракує необхідного заплутування для бажаного основного стану, жоден класичний оптимізатор не зможе досягти цього основного стану.
- Потенційно велика кількість параметрів: дуже виразний ансац може мати так багато параметрів, що ітерації мінімізації стають надзвичайно тривалими.
- Значні витрати на вимірювання: у VQE estimator використовується для оцінки математичного очікування кожного доданку гамільтоніана. Більшість гамільтоніанів, що становлять інтерес, містять доданки, які неможливо оцінити одночасно. Це може зробити VQE ресурсоємним для великих систем зі складними гамільтоніанами[1].
- Вплив шуму: коли класичний оптимізатор шукає мінімум, зашумлені обчислення можуть його збити з пантелику і відвести від справжнього мінімуму або затримати збіжність. Одним із можливих рішень є використання найсучасніших технік мітигації та придушення помилок[2-3] від IBM.
- Безплідні плато: ці регіони зникаючих градієнтів[2-3] існують навіть за відсутності шуму, але шум робить їх ще більш проблематичними, оскільки зміна математичних очікувань через шум може бути більшою, ніж зміна від оновлення параметрів у цих безплідних регіонах.
4.2 Зв'язок з іншими підходами
Adapt-VQE
Алгоритм ADAPT-VQE (Adaptive Derivative-Assembled Pseudo-Trotter Variational Quantum Eigensolver — адаптивний варіаційний квантовий власне-розв'язувач з псевдотроттеризованою збіркою похідних) є вдосконаленням оригінального алгоритму VQE, спрямованим на підвищення ефективності, точності та масштабованості квантових симуляцій, зокрема в квантовій хімії.
Оригінальний алгоритм VQE, описаний упродовж цього уроку, використовує заздалегідь визначений фіксований ансац для наближення основного стану системи. У нашому випадку ми використовували efficient_su2 з одним повторенням і вентилями обертання Y та RZ. Хоча параметри у вентилях RZ змінювались, структура цього ансацу та використовувані вентилі залишалися незмінними.
ADAPT-VQE вирішує обмеження VQE за допомогою адаптивної побудови ансацу. Замість того щоб починати з фіксованого ансацу, ADAPT-VQE динамічно будує ансац ітеративно. На кожному кроці він вибирає оператор із заздалегідь визначеного пулу (наприклад, феріонних операторів збудження), який має найбільший градієнт відносно енергії. Це гарантує додавання лише найбільш впливових операторів, що веде до компактного й ефективного ансацу[4-6]. Такий підхід може мати кілька корисних ефектів:
- Зменшення глибини контуру: завдяки нарощуванню ансацу поступово й зосередженню лише на необхідних операторах ADAPT-VQE мінімізує кількість вентильних операцій порівняно з традиційними підходами VQE[5,7].
- Підвищена точність: адаптивна природа дозволяє ADAPT-VQE відновлювати більше кореляційної енергії на кожному кроці, що робить його особливо ефективним для сильно корельованих систем, з якими традиційний VQE справляється погано[8,9].
- Масштабованість і стійкість до шуму: компактний ансац зменшує накопичення вентильних помилок, скорочує обчислювальні витрати й обмежує кількість варіаційних параметрів, які потрібно мінімізувати.
ADAPT-VQE також не є досконалим. У деяких випадках він може потрапити в локальні мінімуми або сповільнитися через них, а також може страждати від надмірної параметризації. Крім того, він може бути досить ресурсоємним, оскільки вимагає обчислення градієнтів і оптимізації параметрів для багатьох структур вентилів.
Квантова оцінка фази (QPE)
QPE подібний за призначенням до VQE, але дуже відрізняється за реалізацією. QPE вимагає відмовостійких квантових комп'ютерів через загалом глибокі квантові контури та високий рівень когерентності. Коли QPE стане реалізовним, він буде точнішим за VQE. Один із способів описати різницю — через точність як функцію глибини контуру. QPE досягає точності при глибинах контуру, що масштабуються як [10]. VQE потребує вибірок для досягнення тієї самої точності[10,11].
Крайлів, SQD, QSCI та інші методи цього курсу
VQE допоміг утвердити квантові алгоритми, які досі залежать від класичних комп'ютерів — не лише для керування квантовим комп'ютером, а й для суттєвих частин алгоритму. Кілька таких алгоритмів є предметом розгляду в решті цього курсу. Тут ми даємо поверхневий опис деяких з них виключно для порівняння з VQE. Детальніше вони будуть пояснені в наступних уроках.
Квантова діагоналізація Крайлова (KQD)
Методи підпростору Крайлова — це способи проектування матриці на підпростір для зменшення її розмірності й спрощення роботи з нею, зберігаючи при цьому найважливіші властивості. Один із прийомів цього методу полягає у генерації підпростору, що зберігає ці властивості; виявляється, що генерація такого підпростору тісно пов'язана з добре відомим методом на квантових комп'ютерах — тротеризацією.
Існує кілька варіантів квантових методів Крайлова, але загалом підхід такий:
- Використовувати квантовий комп'ютер для генерації підпростору (підпростору Крайлова) за допомогою тротеризації
- Проектувати матрицю інтересу на цей підпростір Крайлова
- Діагоналізувати новий проектований гамільтоніан за допомогою класичного комп'ютера
Квантова діагоналізація на основі вибірки (SQD)
Квантова діагоналізація на основі вибірки (SQD) пов'язана з методом Крайлова тим, що також намагається зменшити розмірність матриці для діагоналізації, зберігаючи ключові властивості. SQD робить це таким чином:
- Починати з початкового наближення для основного стану та підготувати систему в цьому основному стані.
- Використовувати Sampler для вибірки бітових рядків, що складають цей стан.
- Використовувати колекцію обчислювальних базисних станів із sampler як підпростір, на який проектується матриця інтересу.
- Діагоналізувати меншу проектовану матрицю за допомогою класичного комп'ютера.
Це пов'язано з VQE тим, що задіює класичні та квантові обчислення для суттєвих компонентів алгоритму. Обидва методи також поділяють вимогу підготовки хорошого початкового наближення або ансацу. Але розподіл роботи між класичним і квантовим комп'ютерами в SQD більше нагадує метод Крайлова.
Насправді метод Крайлова та SQD нещодавно були об'єднані в метод квантової діагоналізації Крайлова на основі вибірки (SKQD) [12].
Квантова взаємодія конфігурацій у підпросторі
Квантова обрана конфігураційна взаємодія (QSCI)[13] — це алгоритм, що виробляє наближений основний стан гамільтоніана шляхом вибірки пробної хвильової функції для визначення значущих обчислювальних базисних станів з метою генерації підпростору для класичної діагоналізації. І SQD, і QSCI використовують квантовий комп'ютер для побудови зменшеного підпростору. Додаткова перевага QSCI полягає в підготовці стану, особливо в контексті хімічних задач. Він використовує різні стратегії, наприклад стани, еволюціоновані в часі [14], і набір ансаців, натхнених хімією. Зосереджуючись на ефективній підготовці стану, QSCI скорочує квантові обчислювальні витрати для хімічних гамільтоніанів, зберігаючи при цьому високу точність і використовуючи стійкість до шуму від технік квантової вибірки стану [15]. QSCI також надає адаптивну техніку побудови, яка забезпечує більше ансаців для кращого результату.
Типовий робочий процес QSCI для хімічної задачі виглядає так:
- Побудувати молекулярний гамільтоніан за допомогою обраного програмного забезпечення (наприклад, SciPy).
- Підготувати алгоритм QSCI, вибравши відповідний початковий стан і ансац, натхненний хімією, з попередньо обраним набором параметрів.
- Вибрати значущі базисні стани і діагоналізувати гамільтоніан за допомогою класичного комп'ютера для отримання енергії основного стану.
- Часто використовують відновлення конфігурацій [16] і постселекцію симетрії [15] як техніку постобробки.
- За бажанням, робочий процес адаптивного QSCI має додатковий цикл оптимізації від кроку 2 до кроку 3 з використанням більшої кількості ансаців з випадковими початковими станами.
Перевір своє розуміння
Що спільного між VQE і всіма іншими методами, переліченими вище (за винятком QPE, який не описано детально)?
Answer
Всі вони передбачають пробний стан або хвильову функцію певного виду. Всі вони працюють найкраще, коли початкове наближення для цього пробного стану є відмінним.
Інша правильна відповідь: всі вони найлегше реалізуються, коли гамільтоніан легко вимірювати (можна розсортувати у відносно невелику кількість груп комутуючих операторів Паулі).
Чого VQE не має спільного з жодним із інших методів, перелічених вище?
Answer
Класичних оптимізаторів. Жоден з інших методів не використовує алгоритми класичної оптимізації для вибору варіаційних параметрів.
Посилання
[2] https://en.wikipedia.org/wiki/Variational_quantum_eigensolver
[3] https://journals.aps.org/prapplied/abstract/10.1103/PhysRevApplied.19.024047
[4] https://arxiv.org/abs/2111.05176
[6] https://inquanto.quantinuum.com/tutorials/InQ_tut_fe4n2_2.html
[7] https://www.nature.com/articles/s41467-019-10988-2
[8] https://arxiv.org/abs/2210.15438
[9] https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.6.013254
[10] https://arxiv.org/html/2403.09624v1
[11] https://www.nature.com/articles/s42005-023-01312-y
[13] https://arxiv.org/abs/1802.00171
[14] https://arxiv.org/abs/2103.08505
[15] https://arxiv.org/html/2501.09702v1
[16] https://quri-sdk.qunasys.com/docs/examples/quri-algo-vm/qsci/
[17] https://arxiv.org/abs/2412.13839